Adenosine is a small ubiquitous molecule known for its role as an energy and genetic code building block. Adenosine also serves as a ligand for Adenosine receptors, members of the G-protein coupled receptor superfamily and is involved in many physiological processes including regulation of sleep and the level of arousal, neuroprotection, regulation of seizure susceptibility, locomotor effects as well as involved in many pathophysiological conditions. Alomone Labs offers antibodies to all Adenosine receptors, providing an essential tool in the understanding of this system in normal and disease states. Although this system is clearly functional in the periphery, this short review mainly focuses on the role of adenosine and its receptors in the brain.
Introduction
Adenosine is an endogenous nucleoside involved in many biochemical processes and best known for its role in energy transfer in the form of ATP, and the secondary messenger molecule, cAMP, involved in cell signaling pathways. Adenosine in its pure nucleosidic form also influences many functions in the central nervous system (CNS); its levels are determined by the ratio between energy delivery and energy use; thus, increased neuronal activity, particularly hypoxia or ischemia, results in noticeably elevated levels of adenosine32. Adenosine is not stored in vesicles, is not released by exocytosis and does not act only in synapses17, and therefore is not a classical neurotransmitter. As adenosine has a very short half-life and is quite unstable, its effects are usually local26. Adenosine can appear in the extracellular area via three different mechanisms: 1) through nucleoside transporters following an increase in the intracellular levels of adenosine or a reversal of the Na+ gradient; 2) extracellular formation following the release of ATP7,40; 3) extracellular formation following cAMP release11,28. The removal of adenosine from the extracellular space occurs through nucleotide transporters, regulated by the activation of adenosine receptors (ARs) through protein kinase pathways8.
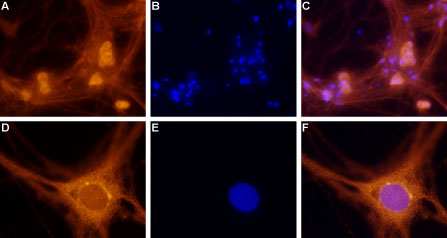
Immunocytochemical staining of paraformaldehyde-fixed and permeabilized rat dorsal root ganglion (DRG) primary culture. A. Staining of DRG cells with Anti-Adenosine A1 Receptor Antibody (#AAR-006), (1:100), followed by goat anti-rabbit-AlexaFluor-555 secondary antibody. B. Nuclear staining of cells using the cell-permeable dye Hoechst 33342. C. Merged images of A and B. F. Merged images of D and E. Magnification: A-C: x20, D-F: x100.
Experimental procedure and figure processed at Alomone Labs.
Adenosine Receptors
Adenosine receptors were defined in the 1970s, partly based on the antagonistic effects of caffeine15. To date, four adenosine receptors have been identified and cloned; Adenosine A1, A2A, and A2B receptors are well conserved among mammals, while adenosine A3 receptor shows considerable structural variability within different species and compared to the other adenosine receptors. Splice variants of all four receptors are also expressed17. The brain expresses high concentrations of adenosine receptors, where adenosine has been shown to be involved in both normal and pathophysiological processes. Due to good pharmacological tools, the distribution of both adenosine A1 and A2A receptors is well known16; A1 receptor shows the highest abundance in the brain, while A2A receptor is expressed at high levels in only a few regions of the brain11. Pharmacological tools available for A2B and A3 receptors are not so extensive and their distribution is mostly based on their corresponding mRNA levels16; both receptors are also expressed in the brain. In fact, the A3 receptor is the only adenosine receptor subtype which was cloned before any pharmacological identification21. Although this short review focuses on adenosine receptors mainly in the brain, it is important to note that they are also distributed in the periphery.
Cell Signaling
The four subtypes of adenosine receptors are G-protein coupled receptors (GPCRs). All four receptors respond to adenosine albeit with different affinities; adenosine A1, A2A, and A3 receptors are considered as high affinity while adenosine A2B receptor requires higher concentrations of adenosine to be activated. Adenosine receptors could also be differentiated on the basis of the signaling pathway(s) they activate or inhibit. A1 and A3 receptors interact with the pertussis toxin-sensitive G-proteins of the Gi/o family13. A3 receptor can also couple Gq thereby also activating phospholipase C, inositol triphosphate and intracellular Ca2+ influx21. A2A and A2B activate adenylyl cyclase through Gs. In addition, A2B receptors can also bind Gq13. While the basic components of the activation of these receptors are the G-protein heteromer, many additional proteins affect the signaling pathways activated in response to adenosine receptor stimulation by direct interaction with the G-proteins and the receptors33. Following activation of the G-proteins, enzymes and some ion channels are affected; for example A1 receptors are thought to activate several types of K+ channels, leading to membrane hyperpolarization23 and inactivate N, P, and Q-type Ca2+ channels1, as well as modulate NMDA receptor function27. On the other hand, A3 receptors activate ATP-sensitive K+ channels (KATP)31. One interesting aspect of adenosine receptor signaling involves their interaction with other types of G-protein coupled receptors. Synergistic interactions have been reported between low concentrations of A1 and metabotropic GABA (B) agonists on inwardly rectifying K+ channels (IRKs)37. There is also extensive evidence from studies on direct interactions between A2A and D1 dopamine receptors, and between A1 and D2 receptors4,22,24 and reviewed in reference 19). In addition to interaction, dimerization between different GPCRs is being increasingly proposed as a common molecular mechanism for cross-talk among various G-protein signaling pathways, and has been observed for many neurotransmitter receptors17. Heteromeric associations were reported for A1 receptor and P2Y1 purinergic receptors39 as well as for A1 and mGluR1 metabotropic glutamate receptor6 and A2A with mGluR514.
Inhibition of Neurotransmitter Release
The most widely recognized effect of adenosine on nerve activity is the inhibition of neurotransmitter release and neuronal excitability via the activation of A1 receptors10,11,18. A1 receptors are more effective in depressing excitatory than inhibitory transmission in the CNS. This inhibition of excitatory transmission largely depends on the activation of presynaptic adenosine A1 receptors which subsequently inhibits the release of glutamate1,3,12.
Ischemia, Neurological and Psychiatric Disorders
The neuroprotective effect of adenosine against brain injury is mostly mediated by adenosine A1 receptor stimulation, as A1 receptor agonists diminish brain damage and the opposite effect is observed using A1 receptor antagonists. Also, activation of A1 has been well documented to protect against ischemic brain injury in adult animal models36.
While A1 activation leads to neuroprotection, activation of A2A may contribute to neuronal injury in several neurological disease models. Pharmacological studies using A2A receptor selective antagonists constantly show reduced ischemic brain damage20,29. It should also be noted though, that in some experimental models, A2A receptor agonists are neuroprotective17. The fact that both A2A receptor agonists and antagonists have protective effects may reflect the complex actions of A2A receptors. A2A receptor antagonists may impose their neuroprotective effect at an early phase of injury (perhaps by decreasing glutamate release) and have opposite effects at the later phase of injury17. The ischemic scenario is similar for that of A3; the acute administration of A3 agonists (before imposing ischemia) demonstrates an increase in ischemic brain damage while a chronic administration has protective effects38.
In the past decade, specific A2A receptor antagonists have emerged as promising pharmacological agents for the treatment of Parkinson’s disease, primarily because of their colocalization with D2 dopamine receptors and the profound antagonistic interaction between adenosine and dopamine systems30. Indeed, continuous work on A2A receptor antagonists as potential treatment for Parkinson’s disease is being done.
Several lines of evidence suggest a potential role of A2A receptors in the pathogenesis of Huntington’s disease. A2A receptors are highly enriched in the striatum, the brain region which is highly degenerated in Huntington’s disease9. Neurochemical studies in a Huntington’s disease mouse model also show that some of the earliest changes are reduced gene expression including that of A2A receptor, suggesting a possible involvement in the pathogenesis of Huntington’s disease5.
A2A receptors may also be involved in Alzheimer’s disease (AD), since acetylcholine release is enhanced by A2A receptor activation. Therefore, increased acetylcholine mediated by A2A receptors may be beneficial for patients with Alzheimer’s disease35. In addition, adenosine A1 receptor expression has been shown to undergo a decrease of 40-60% in the hippocampus of postmortem AD subjects compared to healthy counterparts25,34. However, some areas in the brain also show upregulation of A1 receptors in Alzheimer’s Disease34. The involvement of adenosine A1 receptor in AD is further strengthened by the observations that in degenerative neurons, A1 colocalizes with Aβ-plaques, and phosphorylated tau protein, two well established markers for AD. In addition, in a human neural cell line (SH-SY5Y), Aβ-plaque formation and tau phosphorylation are both mediated by A1 receptor2.
In addition to neurological disorders, adenosine receptors are also marked targets for the treatment of some psychiatric disorders such as schizophrenia as well as some addictive behaviors17.
Alomone Labs has extensively expanded its GPCR portfolio. We offer antibodies to all four adenosine receptors: Anti-Adenosine A1 Receptor Antibody (#AAR-006), Anti-Adenosine A2A Receptor Antibody (#AAR-002), Anti-Adenosine A2B Receptor (extracellular) Antibody (#AAR-003), and Anti-Adenosine A3 Receptor Antibody (#AAR-004). Alomone Labs Adenosine Receptor antibodies enable the detection of their respective receptors using various applications such as western blot, immunohistochemistry, and immunocytochemistry (see the adjoined figures).
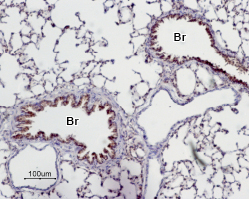
Immunohistochemical staining of paraffin emedded rat lung sections using Anti-Adenosine A2A Receptor Antibody (#AAR-002), (1:50). Adenosine A2A receptor is expressed in the respiratory epithelium of the bronchioli (Br). Note that smooth muscle and endothelium in blood vessels are negative. Hematoxilin is used as the counterstain.
Experimental procedure and figure processed at Alomone Labs.

1,2,5. Anti-Adenosine A2A Receptor Antibody (#AAR-002), (1:200).3,4,6. Anti-Adenosine A2A Receptor Antibody, preincubated with the negative control antigen.
Experimental procedure and figure processed at Alomone Labs.

Immunohistochemical staining of rat lung paraffin embedded sections using Anti-Adenosine A2B Receptor (extracellular) Antibody (#AAR-003), (1:50). Staining is present in the respiratory epithelium of the bronchiole (Br) as well as in the pneumonocytes of the alveolar wall (alveoli, (A)). Color reaction was obtained with SuperPicture HRP-conjugated polymer (Zymed) followed by DAB. Hematoxilin is used as the counterstain.
Experimental procedure and figure processed at Alomone Labs.

1, 3, 5. Anti-Adenosine A2B Receptor (extracellular) Antibody (#AAR-003), (1:200).2, 4, 6. Anti-Adenosine A2B Receptor (extracellular) Antibody, preincubated with the negative control antigen.
Experimental procedure and figure processed at Alomone Labs.

Immunocytochemical staining of paraformaldehyde-fixed and permeabilized human melanoma cells (A2058). A, D. Cells were stained using Anti-Adenosine A3 Receptor Antibody (#AAR-004), (1:100), followed by goat anti rabbit- AlexaFluor-555 secondary antibody. B, E. Nuclear fluorescence staining of cells using the membrane permeable DNA dye Hoechst 33342. C. Merged image of panels A and B. F. Merged image of panels D and E.
Experimental procedure and figure processed at Alomone Labs.
References
- Ambrosio, A.F. et al. (1997) Eur. J. Pharmacol. 340, 301.
- Angulo, E. et al. (2003) Brain Pathol. 13, 440.
- Barrie, A.P. and Nicholls, D.G. (1993) J. Neurochem. 60, 1081.
- Canals, M. et al. (2003) J. Biol. Chem. 278, 46741.
- Cha, J.H. et al. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 6480.
- Ciruela, F. et al. (2001) J. Biol. Chem. 276, 18345.
- Cunha, R.A. (2001) Neurochem. Res. 26, 979.
- Delicado, E.G. et al. (1991) Biochem. J. 279, 651.
- DiFiglia, M. (1990) Trends Neurosci. 13, 286.
- Dunwiddie, T.V. (1985) Int. Rev. Neurobiol. 27, 63.
- Dunwiddie, T.V. and Masino, S.A. (2001) Annu. Rev. Neurosci. 24, 31.
- Fastbom, J. and Fredholm, B.B. (1985) Acta Physiol. Scand. 125, 121.
- Feoktistov, I. and Biaggioni, I. (1997) Pharmacol. Rev. 49, 381.
- Ferre, S. et al. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11940.
- Fredholm, B.B. et al. (1994) Pharmacol. Rev. 46, 143.
- Fredholm, B.B. et al. (2000) Naunyn Schmiedebergs Arch. Pharmacol. 362, 364.
- Fredholm, B.B. et al. (2005) Int. Rev. Neurobiol. 63, 191.
- Fredholm, B.B. and Dunwiddie, T.V. (1988) Trends Pharmacol. Sci. 9, 130.
- Fuxe, K. et al. (1998) Brain Res. Brain Res. Rev. 26, 258.
- Gao, Y. and Phillis, J.W. (1994) Life Sci. 55, PL61.
- Gessi, S. et al. (2008) Pharmacol. Ther. 117, 123.
- Gines, S. et al. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 8606.
- Greene, R.W. and Haas, H.L. (1991) Prog. Neurobiol. 36, 329.
- Hillion, J. et al. (2002) J. Biol. Chem. 277, 18091.
- Kalaria, R.N. et al. (1990) Neurosci. Lett. 118, 257.
- Klinger, M. et al. (2002) Cell Signal. 14, 99.
- Klishin, A. et al. (1995) Neuroscience 65, 947.
- Latini, S. and Pedata, F. (2001) J. Neurochem. 79, 463.
- Monopoli, A. et al. (1998) Neuroreport 9, 3955.
- Morelli, M. and Pinna, A. (2001) Neurol. Sci. 22, 71.
- Mozzicato, S. et al. (2004) FASEB J. 18, 406.
- Newby, A.C. (1991) Adv. Exp. Med. Biol. 309A, 265.
- Pitcher, J.A. et al. (1998) Annu. Rev. Biochem. 67, 653.
- Rahman, A. (2009) Curr. Neuropharmacol. 7, 207.
- Ribeiro, J.A. et al. (2003) Drug News Perspect. 16, 80.
- Rudolphi, K.A. et al. (1992) Cerebrovasc. Brain Metab. Rev. 4, 346.
- Sodickson, D.L. and Bean, B.P. (1998) J. Neurosci. 18, 8153.
- Von Lubitz, D.K. et al. (1994) Eur. J. Pharmacol. 263, 59.
- Yoshioka, K. et al. (2002) FEBS Lett. 531, 299.
- Zimmermann, H. (2000) Naunyn Schmiedebergs Arch. Pharmacol. 362, 299.