Introduction
Cardiac muscle cells (myocytes) are electrically excitable cells, interconnected in groups that respond to stimuli as a unit, contracting together whenever a single cell is stimulated.
Unlike the cells of other muscles and nerves, these cells show a spontaneous, intrinsic rhythm generated by specialized “pacemaker” cells, located in the sinoatrial (SA), and atrioventricular (AV) nodes of the heart. The cardiac cells also have an unusually long action potential, which can be divided into five phases (0 to 4)1,2.
The action potential in the heart
The initial upstroke of the cardiac action potential is determined by the opening and closing of Na+ channels (phase 0).
The configuration and rate of repolarization of action potentials are controlled by many types of K+ channel currents that differ with respect to their kinetics, density of the channels in the plasma membrane and their location in the heart.
Initial repolarization (phase 1) is mediated by the opening of transient outward K+ channels (Ito). This is followed by a plateau (phase 2) that is characterized by high membrane resistance, resulting from the almost equal flow of outward currents through delayed rectifier K+ channels (Ikr, Iks, Ikur) and inward flow of current through L-type Ca2+ channels. The rate of terminal repolarization (phase 3) is enhanced after the plateau phase due to the increasing conductance of the rapid delayed rectifier K+ currents (Ikr) and the inward rectifier K+ current (Ik1). In the last phase (phase 4), a gradual increase in Na+ permeability occurs, depolarizing the membrane towards the threshold of the next action potential1, 2. The contractile machinery of the myocardial cell in response to the action potential is essentially the same as in striated muscle. The force of contraction of the cardiac muscle is directly related to the concentration of free (unbound) cytosolic Ca2+.
Ca2+ comes from two sources: extracellular, where opening of voltage-sensitive calcium channels causes an immediate rise in free cytosolic Ca2+; and intracellular due to the release of Ca2+ from the sarcoplasmic reticulum (SR) and mitochondria. The removal of Ca2+ from the cytosol, which terminates the contraction, can be either via the Na+-Ca2+ exchanger, which reversibly exchanges Ca2+ ions for Na+ ions across the cell membrane or by Ca2+ re-uptake to the SR and the mitochondria (by the PAMCA and the SERCA ATPase pumps)1,3.
The extraordinary biological importance of each of the ion channels and exchangers mentioned above, makes them an important target for specific natural neurotoxins produced by animals and plants featured in this mini review, in addition to the organic compounds utilized today for modulating heart function.
Na+ channel pharmacology
Voltage-gated Na+ channels (VGSCs) are responsible for the initial inward current during the depolarization phase of action potential in excitable cells, and thus important to cardiac and nerve function4-6. This short yet abundant influx of Na+ ions begins a cascade of events resulting in contraction and/or conduction of electrical impulses. A large number of biological neurotoxins exert their toxic effects by modifying the properties of Na+ channels. Pharmacological competition studies and mutagenesis have defined five neurotoxin binding sites on the Na+ channel, (as shown in Table 1)6,7.
Tetrodotoxin citrate (#T-550), isolated from the puffer fish (Tetraodontidae) and Batrachotoxin, isolated from the Colombian arrow frog (Phyllobated), are the most specific VGSC neuromodulators, and their binding is rapidly reversible. The cardiac muscle Na+ channels are less sensitive than those of brain and skeletal muscle. They require micromolar (TTX-resistant) instead of nanomolar (TTX-sensitive) concentration of TTX to be blocked5.
μ-Conotoxin GIIIA (#STC-280) and μ-Conotoxin GIIIB (#C-270) are biologically active peptides, isolated from the venom of the cone snails (Conus geographus). They target VGSCs and cause the blockage of these channels. The skeletal muscle exhibits high affinity to μ-Conotoxins, whereas brain and heart isoforms are resistant to the block5.
Batrachotoxin and Grayanotoxin (from heather plant family), are natural lipid-soluble alkaloid polycyclic toxins, which exert neurotoxic action on nerve and muscle membranes. In heart muscle, they were shown to produce a concentration-dependent reversible positive inotropic effect5.
Type I sea anemone toxins and scorpion α-toxins (isolated from the north African genera scorpions Androctonus, Buthus and Leiurus) were first discovered as polypeptide cardiostimulants.
The type I sea anemone toxins activates VSGCs by slowing the inactivation and includes: ATX-I, ATX-II8,9 and ATX-V (isolated from Anemonia sulcata) and Anthopleurin-A, Anthopleurin-B, Anthopleurin-C (APE 2-1)10,11 and its close homolog, APE 1-211 (isolated from Anthopleura elegantisima). δ-Atracotoxins (δ-Atracotoxin-Ar1 and δ-Atracotoxin-Hv1 isolated from the Australian funnel-web spiders family, Atracinae) are a relatively new class of toxins, which also slow the inactivation of VGSCs5,12.
Scorpion β-toxins (isolated from the venom of North and South American scorpions from the genus Centuroides) shift the voltage dependence of activation to more negative membrane potentials, thereby enhancing channel activation5.

K+ channel pharmacology
The configuration of cardiac action potential varies considerably from one region of the heart to another. These differences are caused by differential cellular expression of several types of K+ channel genes. Channels encoded by these genes can be grouped into several classes depending on the stimulus that permits the channels to open and conduct K+ ions1. K+ channels are activated by changes in transmembrane voltage or by binding of ligands. Voltage-gated channels are generally the most important players in determining the shape and duration of action potentials and include the delayed rectifiers and the transient outward K+ channels.
Ligand-gated channels include those that probably have only minor roles in shaping repolarization under normal conditions1. Inward rectifier K+ channels are unique in that they are generally kept in the open state but can be blocked in a voltage-dependent manner. Other K+ channels have been described that provide a small background leak conductance1. Table 2 lists only those channels in the heart for which there are specific known neurotoxins, isolated from animals or plants. There are also other K+ channels in the heart, for which specific neurotoxins are not yet known, including: voltage-gated K+ channels, Shaker (Kv1.4 and Kv1.5), MinK (IsK), KvLQT1, the Inward Rectifier (Kir2.1), Sulfonylurea receptor (SUR2), the two-pore K+ channels (TWIK1, TASK-1) and the hyperpolarization activated cation channels (HCN2 and HCN4).
Dendrotoxins are a family of homologous peptides isolated from green and black mamba venoms (Dendroaspis sp.). α-Dendrotoxin and Dendrotoxin-I (#D-390) are potent blockers of the Kv1.2 K+ channel13-15.
Tityustoxin-Kα (#STT-360) (isolated from the Brazilian scorpion Tityus serrulatus) blocks the Kv1.2 K+channel16, 17.
Hanatoxin 1 and Hanatoxin 2 are two peptides, isolated from the Chilean tarantula , (Grammostola spatulata). They specifically inhibit the Kv2.1 channel, a Shab-related K+ channel, with little effect on Shal-related K+ channel and with no effect on Shaker-related, Shaw-related and EAG K+ channels18. Tertiapin (#STT-250) (isolated from the honey bee venom, Apis mellifera) is a peptidyl toxin, which blocks Kir3.1 and Kir3.4, and other types of Kir K+ channels19,20. Recently it was also found that Tertiapin selectively inhibits muscarinic K+ channels (KAch) in cardiac myocytes21.
Heteropodatoxins (isolated from the venom of Heteropda venatoria spider) includes three peptide toxins (Heteropodatoxins 1, 2, and 3) that block the Kv4.2 K+ channel and thus prolong cardiac action potentials22.
Phrixotoxin 1 and Phrixotoxin 2 are two new toxins that have been isolated from the Chilean fire tarantula (Phrixotrichus auratus) and specifically block Kv4.3 and Kv4.2 currents that underlie the It0123.
Ergtoxins are a new class of peptides, which block ERG K+ channels of nerve, heart and endocrine cells. The ERG channels (ether-a-go-go) are crucial mainly in human long-QT syndrom and arrythmia24-26. Ergtoxin is isolated from the venom of a Mexican scorpion (Centruroides noxius). It specifically inhibits ERG channels of different species and distinct histogenesis, but does not block the closely related EAG channels24,25.
Another toxin which is included in this group and isolated from a central Asian scorpion (Buthus eupeus), named BeKm-1, was found to be a selective high affinity and potent blocker of hERG1 channels27, 28.

Ca2+ signaling pharmacology
Ca2+ channels are critical to normal cardiac function. They are involved in the generation and conduction of the action potential and in contraction.
Heart cells contain at least five types of Ca2+ currents, three of which (L-type, T-type, and the newly described current (ICa TTX)) are expressed on the surface membrane, and two (the ryanodine receptor, RYR, and the IP3receptor expressed on the sarcoplasmic reticulum) in internal membranes3.
The ATPase pumps in the plasma membrane (PAMCA) and i n the sarco/endopl asmic reticulum (SERCA) are responsible for preventing the intracellular Ca2+ from reaching high cytotoxic levels in the intracellular space. The L-type Ca2+ channel is the most abundant Ca2+ channel in the heart and it is responsible for Ca2+ entry into the cell and triggers contraction. The T-type Ca2+ channels are most prevalent in the conduction system and are probably involved in automaticity3.
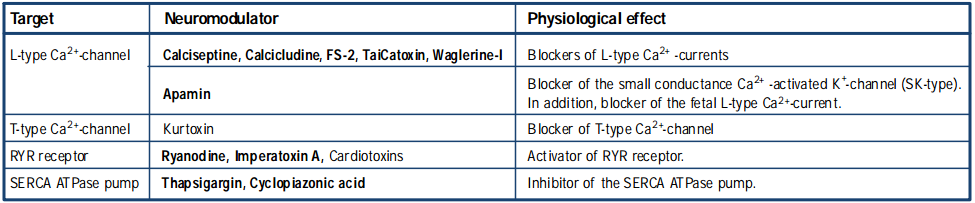
Several neurotoxins which block the L-type Ca2+ channels exist and include: Calciseptine (#C-500)29, Calcicludine (#C-650, replaced with #SPC-650)30 and FS-2 (#F-700)31 (isolated from the mamba snake venoms), TaiCatoxin32, isolated from Oxyuranus s. scuttelatus and Waglerine- I33, isolated from Trimeresurus wagleri. Apamin (#STA-200) (isolated from the honey bee, Apis Mellifera, venom) is known as a blocker of the small conductance Ca2+-activated K+ channel (SK-type)13. In addition, it was also shown to act as a highly potent fetal L-type Ca2+ current blocker in single heart cells, without affecting the T- type Ca2+current34-36. Only recently, a new T-type Ca2+ channel blocker was discovered, Kurtoxin, which is isolated from the venom of a South African scorpion (Parabuthus transvaalicus). This toxin was found to specifically bind to the α1G T-type Ca2+ channel with high affinity and inhibits the channel by modifying voltage-dependent gating. Kurtoxin was also found to interact with voltagegated Na+ channels and slows their inactivation37.
The receptors expressed on the sarcoplasmic reticulum are responsible for activating a variety of Ca2+channels, in order to increase the intracellular Ca2+ level. The RYR receptor is activated by Ryanodine (#R-500), which is a natural product agonist (isolated from Ryania speciosa vahl plant)38 and by Imperatoxin A (#I-300) (isolated from the venom of the scorpion Pandinus imperator)39. The SERCA ATPase pump is selectively inhibited by Thapsigargin (#T-650)40, 41 (isolated from Thapsia garganica) and by Cyclopiazonic Acid (#C-750)42 (isolated from Penicillium cyclopium).
Cardiotoxins are a family of neurotoxins, isolated from cobra snake venoms43 ,44. Several cardiotoxins isolated from the Taiwan cobra (Naja naja atra) venom are known as openers of the ryanodine receptor and alter the activity of the Ca2+/Mg2+-ATPase, thus elevating the intracellular Ca2+ level45,46.
Stretch-Activated Ion Channels (SACs)
Stretch-activated ion channels are ubiquitous and have been studied in a variety of cardiac tissues, implicating a role in the mechanical sensitivity of the heart and in atrial fibrillation. Recently, a new toxin named GsMtx-4, isolated from a tarantula spider (Grammostola spatulata) was found to specifically block cationic stretch activated ion channels and successfully blocked atrial fibrillation in rabbit hearts47,48.
Summary
Nature provides us with a rich source of potent neurotoxins, which modulate the structure and function of ion channels, exchangers and receptors, crucial to normal heart function.
Alomone Labs has always dedicated itself to the discovery of new pharmacological tools, which are specific modulators of ion channels and receptors. Some of these new tools are directed towards channels and receptors, which do not have any pharmacological modulators.
In addition, we concentrate on commercial manufacturing of natural and recombinant neurotoxins.
We plan to make these compounds available to the research community so that more laboratories can make use of these tools. We hope that our efforts will result in helping advance this important field by providing the scientific community with unique, top quality, bioactive compounds.
References
- Tristani-Firouzi, M. et al. (2001) Am. J. of Med. 110, 50.
- Grant, A.O. (2001) Am. J. Med. 110, 296.
- Shorofsky, S.R. and Balke, C.W. (2001) Am. J. Med. 110, 127.
- Catterall, W.A. (2000) Neuron 26, 13.
- Denac, H. et al. (2000) Naunyn-Schmiedebergís Arch pharmacol. 362, 453.
- Marban, E. et al. (1998) J. of Physiol. 508, 647.
- Catterall, W.A. (1980) Ann. Rev. Pharmacol. Toxicol. 20, 15.
- el-Sherif, N. et al. (1992) Circ. Res. 70, 285.
- Boutjdir M. et al. (1996) J. Cardiovasc. Pharmacol. Ther. 1, 149.
- Norton, T.R. (1981) Fed. Proc. 40, 21.
- Bruhn, T. et al. (2001) Toxicon 39, 693.
- Little, M.J. et al. (1998) FEBS Letters 439, 246.
- Shieh, C.C. et al. (2000) Pharmacol. Rev. 52, 557.
- Baker, M. et al. (1993) J. Physiol. 464, 321.
- Harvey, A.L. (2001) Toxicon, 39, 15.
- Werkman, T.R. et al. (1993) Mol. Pharmacol. 44, 430.
- Rogowski R.S. et al. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 1475.
- Swartz, K.J. and MacKinnon, R. (1995) Neuron 15, 941.
- Jin, W. and Lu, Z. (1998) Biochemistry 38, 13291.
- Matsushita, K. et al. (2000) The Japanese J. of Pharmacol. 82, Supplement 1, 130P.
- Kitamura, H. et al. (2000) J. Pharmacol. Exp. Ther. 293, 196.
- Sanguinetti, M.C. et al. (1997) Mol. Pharmacol. 51, 491.
- Diochot, S. et al. (1999) Br. J. of Pharmacol. 126, 251.
- Gurrola, G.B. et al. (1999) FASEB J. 13, 953.
- Bottiglieri, C. et al. (2000) FEBS Letters 479, 155.
- Curran, M.E. et al. (1995) Cell 80, 795.
- Filippov, A.K. et al. (1996) FEBS Letters 384, 277.
- Korolkova, Y.V. et al. (2001) J. Bio. Chem. 276, 9868.
- de Weille, J.R. et al. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 2437.
- Stotz, S.C. et al. (2000) J. Membr. Biol. 174, 157.
- Yasuda, O. et al. (1994) Artery 21, 287.
- Possani, L.D. et al. (1992) Toxicon 30, 1343.
- McArdle, J.J. et al. (1992) Soc. Neurosci. Abstr. 18, 969.
- Bkaily, G. et al. (1992) Am. J. Physiol. 262, H463.
- Bkaily, G. et al. (1985) Am. J. Physiol. 248, H961.
- Schetz, J.A. and Anderson, P.A. (1995) Cardiovasc. Res. 30, 755.
- Chuang, R.S. et al. (1998) Nat. Neurosci. 1, 668.
- Messner, G. (1986) J. Biol. Chem. 261, 6300.
- Tripathy, A. et al. (1998) J. Gen. Physiol. 111, 679.
- Treiman, M. et al. (1998) Trends Pharmacol. Sci. 19, 131.
- Mackiewicz, U. et al. (2000) J. Physiol. Pharmacol. 51, 777.
- Demaurex, N. et al. (1992) J. Biol. Chem. 267, 2318.
- Chang, L.S. et al. (2000) Toxicon 38, 1065.
- Kumar, T.K. et al. (1997) J. Biomol. Struct. Dyn. 15, 431.
- Fletcher, J.E. and Jiang, M.S. (1993) Toxicon 31, 669.
- Wang, H.X. et al. (1999) Clin. Exp. Pharmacol. Physiol. 26, 835.
- Suchyna, T.M. et al. (2000) J. Gen. Physiol. 115, 583.
- Bode, F. et al. (2001) Nature 409, 35.