The immune response is a tightly regulated process, in which any breach of this strict regulation can lead to pathological phenomena. In addition to the more familiar members of the immune response, a key role for ion channels in immune function is now apparent in the research. Here we discuss the prominent involvement of the K+ channels KV1.3 and KCa3.1 (IKCa1) and the purinergic receptor P2X7 in the regulation of key elements of the immune response.
The unexpected role of ion channels
Although not readily apparent, there are more than a few similarities between the immune system and the central nervous system (CNS). Both systems can process complex and dynamic input signals and produce an appropriate and timely response. And both systems use exquisitely complex mechanisms that ensure that the response will match the input signal and thus prevent potential damage. Therefore, it is fitting that immunologists are now using the term “synapse” (or “immunological synapse”) to describe the close encounter and interaction between an antigen-presenting cell (APC) and a T lymphocyte cell.
However, the cells of the immune system were originally categorized as “electrically non-excitable”, that is cells that do not have action potentials generated by voltage-gated ion channels, the hallmark of CNS cells. It is now well established that immune system cells do express a remarkable array of voltage-gated and other types of ion channels1. It appears that ion channels play a prominent role in the regulation of the immune response, much as they do in the context of the CNS.
Regulation of T-cell activation
The KV1.3 and KCa3.1 channels play a key role in T-cell activation. KV1.3 belongs to the Shaker family of voltage-dependent K+ channels. The channel is expressed in various populations of hematopoietic origin such as T and B lymphocytes, macrophages, and natural killer (NK) cells2. The functional channel in T lymphocytes is composed of four KV1.3 subunits and is responsible for maintaining the membrane potential in the resting cell. The KCa3.1 channel (also known as IKCa1 or SK4) is a Ca2+-dependent K+ channel with intermediate conductance. The channel is not voltage-sensitive and opens with submicromolar concentrations of intracellular Ca2+. The channel is tightly bound to calmodulin, its Ca2+sensor, via its C-terminus3. KCa3.1 is expressed at low levels in resting T cells, although it is dramatically upregulated once the T cells have been fully activated4,5.
An adaptive immune response against a specific pathogen takes place when a T helper (TH) lymphocyte becomes engaged or activated through its unique T cell receptor (TCR) (see figure 1). The activated T cell will then proliferate and subsequently differentiate into cell subsets (or effectors) that are best suited to deal with the specific pathogen. The kind of immune response evoked by the invading pathogen can be divided into two categories: a cell-mediated (also called Th1) response primarily directed against intracellular pathogens and viruses or an antibody-dependent (or Th2) response (by way of antibody-producing B lymphocytes) directed primarily against bacteria and parasites. Later still, the activated T cell differentiates into a memory cell that would provide lingering protection against recurrent infections with the same pathogen. Thus, the initial encounter between the APC and the T cell is crucial for the outcome of the immune response.
The “decision” made by the T cell, whether to respond or not and how to respond, is a very complex process regulated by many factors, such as the nature of the antigen, the duration of the encounter with the APC, and the prevailing microenvironment (cytokines, chemokines, etc.). It is precisely at this critical stage that the K+ channels KV1.3 and KCa3.1 are believed to be involved. The current model for understanding the involvement of K+ channels in T cell activation implies that these channels indirectly control the intracellular Ca2+ signals that are indispensable to T cell proliferation.
The earliest step following antigen-induced TCR activation is the tyrosine phosphorylation of several proteins such as Lck and PLCγ1. The latter will induce Ca2+ release from intracellular stores through the production of Inositol 1,4,5-triphosphate (IP3) and subsequently a sustained Ca2+ entry from the extracellular space. It is well known that elevated levels of intracellular Ca2+ must be maintained for a relatively long time (a few hours) to allow the activated T cell to proliferate. Extracellular Ca2+ enters the cell based on its electrical driving force and therefore depolarizes the cell membrane. This induces the activation of the KV1.3 channel which, via K+ efflux, helps to maintain a negative membrane potential, thus allowing for continuous Ca2+ entry. As mentioned above, T cell activation enhances the expression of KCa3.1 channels, which open in response to the increased cytosolic Ca2+ and thus further hyperpolarize the membrane potential6,7.
In summary, the concerted action of both KV1.3 and KCa3.1 allows the sustained rise in intracellular Ca2+ that permits the Ca2+-dependent activation of calcineurin and the transcription factor NF-AT. Consistent with this model, blockers of K+ channels such as Charybdotoxin and Margatoxin effectively inhibit antigen-dependent T cell activation and IL-2 secretion8,9,10. Additionally, specific inhibitors of KCa3.1 were able to inhibit the activation of previously activated T cells, again, consistent with the model in which the increased expression of these channels after activation plays a significant role in a subsequent antigen challenge11.
Moreover, KV1.3 channel inhibitors were able to inhibit T cell-mediated immune responses in vivo, such as delayed-type hypersensitivity or antibody response to foreign antigens12. These results prompted the notion that K+ channel blockers could be used as effective immunosuppressants. Neuroinflammation, for instance, is a major contributor to multiple neurological disorders and driven by activation of microglia. In a mouse model of selective innate immunity, Kv1.3 is required for proinflammatory activation of microglia, and thus Kv1.3 blockers may be valuable therapeutic candidates where microglia-mediated neurotoxicity plays a role in the pathogenesis13.
Although a great deal of progress has been made in the last few years regarding the involvement of K+ channels in general, in the immune response, and in particular the involvement of KV1.3 and KCa3.1, it seems that there is a lot yet to be learned. For instance, one study suggested that the oligomeric composition of the main KV channel might vary in different functional lymphocyte populations. For example, anergized T cells (live cells that are unable to proliferate following stimulation) may express KV1.2, KV1.1, or KV1.6 along with KV1.3, indicating that the substantial physiological and functional changes undergone by a responding T cell may be paralleled by changes in the subunit composition and/or function of the KV channel14. Consistent with this idea, a report showed that exposure of T cells to hypoxia (low oxygen availability) downregulates the KV1.3 channel at the protein level, an effect that could account for the previously described inhibition of T cell proliferation under hypoxic conditions15.
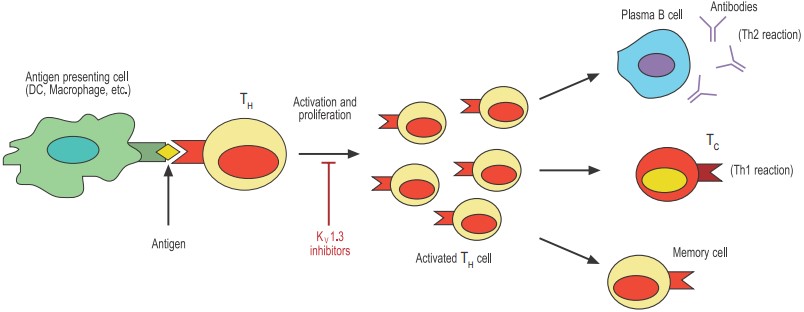
Antigen-presenting cells such as dendritic cells (DCs) and macrophages phagocytose and digest invading pathogens such as bacteria. Selected peptides of the pathogen (antigens) are “presented” to T helper (TH) cells. Only the TH with the appropriate T cell receptor can proliferate and become activated. The activated TH can now stimulate effector mechanisms such as plasma cell-secreted antibodies and T cytotoxic cells (TC) that will help contain the invading pathogen. Finally, the activated TH either dies by apoptosis or differentiates to long-lived memory cells.
Controlling inflammation: The central role of P2X7
The P2X7 receptor is a member of a large family of ligand-gated ion channels that open in response to extracellular ATP. The family comprises 7 members (P2X1-7), which share between 40–50% identity in their amino acid sequence. Activation of all the P2X receptors leads to increased membrane permeability to small cations (Na+, K+, and Ca2+), while sustained stimulation of the P2X7 receptor leads to the generation of an increasingly large membrane pore that is permeable to low-molecular-weight organic cations16. The functional P2X channel is a multimer, but in contrast to the other P2X subunits, P2X7 is unable to form heteromeric channels16. Another unusual characteristic of the P2X7 receptor is that it requires very high ATP concentrations (in the range of millimolar) to become active. This prompted the speculation that there may be another endogenous ligand for the P2X7 receptor.
The P2X7 receptor is widely expressed throughout the hematopoietic lineage. The receptor has been found in T and B cells, dendritic cells (DCs), macrophages, and mast cells and has been implicated in several physiological activities. Although inflammation is often alluded to in the context of pathological conditions such as asthma and atherosclerosis, it is in fact an essential component of a complex mechanism designed to deal with tissue injury and infection. Inflammation often starts when tissue injury (caused for example by invading bacteria) initiates the secretion of chemical signals that recruit different cell populations such as neutrophils, mast cells, dendritic cells, and macrophages. These cells constitute the first line of defense and will on one hand attack the invading bacteria by phagocytosis (macrophages and neutrophils) and on the other hand enlist the cells that are responsible for the permanent eradication of the pathogen (T cells). The P2X7 receptor that is expressed on all the cells involved in the inflammatory process has been identified as a key player in the regulation of inflammation (see Figure 2).
One of the most surprising roles of P2X7 in the immune system is its key role in the secretion of the biologically active interleukin 1β (IL-1β). IL-1β is often regarded as the master proinflammatory cytokine based on its ability to initiate a wide variety of proinflammatory responses from different cell types, ranging from upregulation of metalloproteinases to secretion of IL-617. IL-1β is abundantly expressed by macrophages that have been activated, for example with bacterial LPS present in the sites of infection. However, activated macrophages produce only the inactive propolypeptide form that must be cleaved by caspase-1 before it becomes active. Remarkably, activated macrophages need a second stimulus (independent of LPS) to activate caspase-1 and therefore produce mature IL-1β. One of the most powerful signals that operate in this context is extracellular ATP through the P2X7 receptor18.
The central role of P2X7 in ATP-mediated IL-1β maturation was demonstrated in macrophages isolated from P2X7 knockout mice. These cells failed to produce mature IL-1β even though they produced high quantities of proIL-1β in response to LPS19. Other known functions of P2X7 in the immune response include shedding of L-selectin in leukocytes and lymphocytes, ATP-mediated proliferation and apoptosis, killing of intracellular bacteria, and activation of intracellular inflammatory-related signal transduction pathways such as translocation of NF- B to the nucleus or upregulation of cyclooxygenase-2 (COX-2) in monocytes and macrophages20,21,22. Indeed, mice deficient in P2X7 (P2X7 knockouts) showed a marked reduction of disease severity in a mouse model of inflammatory arthritis, prompting the suggestion that P2X7 can be an attractive target for anti-inflammatory drugs23. Despite all this, it was difficult to reconcile these findings with the fact that very high concentrations of extracellular ATP are necessary to activate P2X7 since these ATP concentrations seemed unphysiological. One particular study might help to dispel the mystery. The authors showed that nicotinamide adenine dinucleotide (NAD) functions as a substrate for ADP-ribosyltransferase 2 (ART2), an ectoenzyme that catalyzes the linkage of ADP-ribose to an acceptor protein, in this case P2X724. ART2-mediated ADP-ribosylation can activate P2X7 and all the known downstream signaling, such as Ca2+mobilization and L-selectin shedding in the absence of ATP. For these effects to take place, only relatively small concentrations of extracellular NAD were required (in the micromolar range). Thus, a putative model for the physiological activation of P2X7 is that extracellular NAD (as extracellular ATP) that is released at the sites of inflammation by the lysis of dying cells, is catabolized by ART2 expressed on the cell membrane of inflammatory cells attracted to the site, and therefore activates the P2X7 channel on those cells.
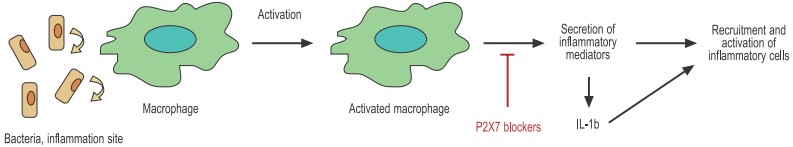
Bacteria activate macrophages through specific receptors in the cell membrane. The activated macrophage secretes a great number of inflammatory mediators including chemokines, cytokines, and others that help recruit and activate other immune cells.
Other functions, other channels
The above account of the involvement of KV1.3, KCa3.1, and P2X7 in the modulation of the immune response by no means summarizes the contribution of ion channels in general to the immune response. Rather, there are many ion channels whose involvement has already been observed and whose function is no less central to the immune response.
The primary example is Ca2+ channels. As mentioned above, a rise in intracellular Ca2+ concentration is crucial to the activation of lymphocytes (both T and B cells) and leukocytes (macrophages, neutrophils, mast cells). The release of Ca2+ from intracellular stores following stimulation is well characterized. However, the molecular identity of the channel that mediates the subsequent Ca2+ entry from the extracellular medium, which is responsible for the majority of the elevated Ca2+ levels, is still elusive. The uncertainty starts with the term used to describe the plasma membrane Ca2+ channel. The names Ca2+ release-activated Ca2+ channels (CRAC) and store-operated Ca2+ channel (SOCC) are used interchangeably, although there may be differences in the biophysical properties of both channels. The leading candidates for the molecular identity of the CRAC channels are members of the TRP gene superfamily. TRPC3 and TRPC6 have been studied in T cells, although it appears that the current favorite is TRPV6 (CaT1)25,26.
Cl– channels have also been observed in lymphocytes where they function to decrease the volume of the cell following osmotic stress1. In addition, blockers of Cl- channels have been shown to inhibit lymphocyte activation and proliferation27. As in the case of the CRAC channels, the molecular identity of the Cl– channels is not clear, although mRNA from the CLC3 channel has been detected in several hematopoietic cell lines28.
Finally, several ion channels have been identified in cells of the immune system but whose function has not yet been thoroughly investigated. This group includes epithelial Na+ channels (ENaC) in lymphocytes29, the water channel aquaporin 9 in neutrophils30, and the nicotinic acetylcholine receptor in B lymphocytes31,32.
Undoubtedly, in the next few years, further studies will reveal new and perhaps not so unexpected roles of ion channels in the immune response.
References
- Feske, S. et al. (2018) Annu. Rev. Immunol. 33, 291.
- Lewis, R.S. et al. (1995) Annu. Rev. Immunol. 13, 623.
- Logsdon, N.J. et al. (1997) J. Biol. Chem. 272, 32723.
- Ghanshani, S. et al. (2000) J. Biol. Chem. 275, 37137.
- Khanna, R. et al. (1999) J. Biol. Chem. 274, 14838.
- Cahalan, M.D. et al. (1997) Curr. Opin. Biotech. 8, 748.
- Matko, J. (2003) Trends Pharmacol. 24, 385.
- Lin, S.C. et al. (1993) J. Exp. Med. 177, 637.
- Leonard, R. J. et al. (1992) Proc. Natl. Acad. Sci. USA 89, 10094.
- Kerschbaum, H.H. et al. (1997) J. Immunol. 159, 1628.
- Wulff, H. et al. (2000) Proc. Natl. Acad. Sci. USA 97, 8151.
- Koo, G.C. et al. (1997) J. Immunol. 158, 5120.
- Lucente, J. et al. (2019) Glia 66, 1881.
- Liu, Q.H. et al. (2002) J. Exp. Med. 196, 897.
- Conforti, L. et al. (2003) J. Immunol. 170, 695.
- North, A.D. et al. (2002) Physiol. Rev. 82, 1013.
- Bent, R. et al. (2018) Int. J. Mol. Sci. 19, 2155.
- Perregaux, D.G. et al. (2000) J. Immunol. 165, 4615.
- Solle, M. et al. (2001) J. Biol. Chem. 276, 125.
- Fairbairn, I.P. et al. (2001) J. Immunol. 167, 3300.
- Budagian, V. et al. (2003) J. Biol. Chem. 278, 1549.
- Jamieson, G.P. et al. (1996) J. Cell Physiol. 166, 637.
- Labasi, J.M. et al. (2002) J. Immunol. 168, 6436.
- Seman, M. et al. (2003) Immunity 19, 571.
- Grafton, G. et al. (2001) Immunology 104, 119.
- Vassilieva, I. O. et al. (2013) J. Membr. Biol. 246, 131.
- Phipps, D.J. et al. (1996) Cell Signaling 8, 141.
- Jiang, B. et al. (2002) Life Sciences 70, 1383.
- Bubien, J.K. et al. (2001) J. Biol. Chem. 276, 8557.
- Loitto, V.M. et al. (2002) J. Leukoc. Biol. 71, 212.
- Skok, M.V. et al. (2003) Mol. Pharmacol. 64, 885.
- Fujii, T. et al. (2017) J. Pharmacol. Sci. 134, 1.