Metabotropic glutamate receptors (mGluRs) are functionally important for inducing long term potentiation (LTP) and long term depression (LTD). They are essential for synaptic transmission, neuronal development, neuroprotection and cognition. Alomone Labs offers antibodies targeting extracellular epitopes of mGluRs. Such antibodies enable to detect mGluRs in living cells, and to monitor cell surface receptors. Also available are fluorescent labeled antibodies which enable colocalization studies involving mGluRs. In this brief report, we summarize papers elegantly using Alomone Labs mGluR antibodies.
Introduction
L-glutamate is the most abundant excitatory neurotransmitter in synapses of the vertebrate central nervous system (CNS)5,9,14,15. It is secreted from presynaptic vesicles as a consequence of intracellular Ca2+accumulation by N- and P/Q-type voltage-gated Ca2+ channels. Upon release, glutamate acts on both ionotrophic receptors (iGluRs; AMPA, NMDA and kainate) which conduct fast excitatory transmission, and metabotrophic, G protein-coupled receptors on postsynaptic membranes, which modulate the plasticity and dynamics of excitable synapses5,8,14. The 8 known mGluRs (mGluR1-mGluR8) share the molecular morphology of G protein coupled receptors (GPCRs): seven transmembrane domains, an extracellular N-terminus, and an intracellular C-terminus, and are grouped into 3 separate classes (I-III) based on their second-messenger partnership and pharmacological profile7,8,12.
Group I
Group I mGluRs, mGluR1 and mGluR5, increase neuronal firing and synaptic transmission by producing the second messengers diacylglycerol and inositol trisphophate from phospholipase C, through Gq protein (of the G protein family) coupled activation7. This leads to the escape of Ca2+ from intracellular stores, and in the activation of protein kinase C (PKC)5,7,14. In turn, activated PKC phosphorylates the C-terminal of group I mGluRs at serine 901. E3 ligase seven in absentia homolog (Siah)-1A then acts to degrade the receptors, by competing with calmodulin (CaM) which regulates their surface expression. Indeed, HeLa cells transfected with mGluR5 and co-expressed with Siah-1A showed almost no surface expression of mGluR5 in immunostaining assays, as opposed to cells expressing mGluR5 that is mutated at threonine 913, a site which is critical for Siah-1A binding (but not of CaM)5. In addition, rat primary hippocampal neurons transfected with Siah-1A also demonstrated decreased mGluR5 cell surface expression as shown in immunocytochemical staining using Anti-mGluR5 (extracellular) antibody (#AGC-007), (Figure 1).
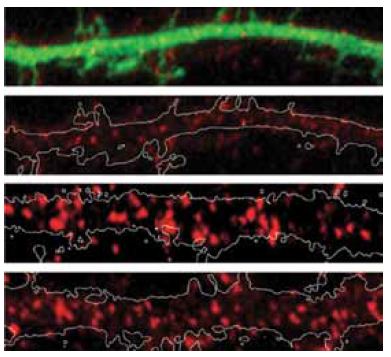
Immunocytochemical staining of rat primary hippocampal neurons transfected with EGFP (Control) or with Siah-1A-EGFP (Siah-1A). Extracellular staining of cells with Anti-mGluR5 (extracellular) antibody (#AGC-007) (red) shows that mGluR5 staining intensities decrease in Siah-1A transfected cells.
Adapted from reference 5 with permission of the Society for Neuroscience.
The polymerization of soluble amyloid β oligomers (Aβo) significantly correlates with cognition disturbances and is putatively linked to the memory loss seen in early stages of Alzheimer’s disease (AD) where Aβos disturb synaptic plasticity, impair long term potentiation (LTP) and enhance long term depression (LTD). Specifically, Aβos contribute to the pathological remodeling of the post synaptic density (PSD) construct in dendritic spines – a meshwork of scaffold proteins and enzymes situated in the excitatory postsynaptic region; notably Homer1b which also interacts with mGluR111. The interaction occurs through Homer1b’s N-terminal EVH1 domain and Shank1, a cytoplasmic PSD protein that bridges the cytoskeleton with various glutamate receptors. It seems that Aβo depletes Homer1b levels which ultimately leads to a decrease in mGluR1 surface levels as shown using Anti-mGluR1 (extracellular) antibody (#AGC-006) in immunocytochemical staining of rat cortical neurons11 (Figure 2). A study investigating the initial effects of Aβo showed that oligomers bind at the membrane surface probably via mGluR5 and limit the GPCR from lateral movement and cause it to cluster at synapses. This subsequently leads to synaptic failure involving in part intracellular Ca2+ overload. Colocalization between Aβo and mGluR5 was observed in living rat hippocampal neurons labeled extracellularly with biotinylated Aβo and Anti-mGluR5 (extracellular) antibody10 (Figure 3).
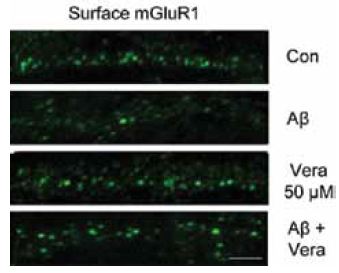
Immunocytochemical staining of rat cortical neurons treated with Aβo. Extracellular staining of cells using Anti-mGluR1 (extracellular) antibody (#AGC-006) (green) shows that mGluR1 cell surface expression significantly decreases upon Aβo treatment.
Adapted from reference 11 with kind permission of Dr. Roselli, F. (from Max-Planck Institute of Psychiatry, Munich, Germany and Department of Neurological and Psychiatric Sciences, University of Bari, Bari, Italy) and of Dr. Almeida, O.F.X. (from Max-Planck Institute of Psychiatry, Munich, Germany).
Cocaine intake can induce cue-associated learning in amygdalar regions, reflected as a dose-dependent craving in response to environmental renditions in the absence of the drug. Group I mGluRs are important neuroplasticity modulators. They are well known mediators of LTP and LTD and are commonly used to investigate neuroadaptivity6,13. In a study using conditioned place preference (CPP), a method used to monitor drug-cue association, the two isoforms of phospholipase D (PLD), PLD1 and PLD2 were coimmunoprecipitated with mGluR1 and mGluR5 from the rat amygdala following cocaine administration using mGluR1 and mGluR5 respective Alomone Labs antibodies6. Furthermore, LTP induced by activating dopaminergic receptors has been found to be PLD-linked and mGluRdependent. As a result, PLD is increased in the conditioned amygdala6. The biological significance of PLD1 and PLD2 regarding cocaine and cue association needs to be further investigated. In a related study, cocaine withdrawal was found to reduce group I mGluR1-mediated LTP via a decrease in inhibitory GABAergic signaling. However, the LTP decrease is not a result of a decrease in protein levels, since western blot analysis of mGluR1 and mGluR5 using Anti-mGluR1 (extracellular) and Anti-mGluR5 (extracellular) antibodies detected similar levels of protein in control groups and groups going through withdrawal13.
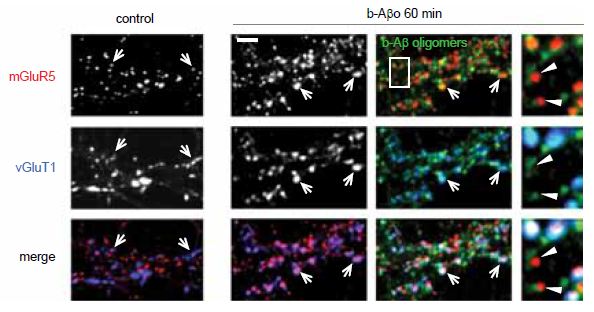
Immunocytochemical staining of rat hippocampal neurons treated with Aβo (b-Aβo). Extracellular staining of cells using Anti-mGluR1 (extracellular) antibody (#AGC-006) demonstrates that mGluR5 cluster following Aβo treatment.
Adapted from reference 10 with permission of Elsevier.
Alomone Labs Antibodies targeting group I mGluRs were also used to find a substantially high level of mGluR5 in dorsal root ganglia (DRG) of diabetic rats7, and to verify the expression of both mGluR1 and mGluR5 in the ventral respiratory group (VRG) island, a region containing the pre-BÖtzinger complex which generates the respiratory rhythm2. Anti-mGluR5 (extracellular) antibody was used to monitor mGluR5 surface expression in immunocytochemical staining of mouse corticostriatal cocultures of wild type and Sapap3-/- mice. SAPAP3 is a postsynaptic protein unique to excitatory synapses and believed to regulate metabotropic glutamate receptors or the signaling pathways they activate. Indeed, mGluR5 expression was significantly higher in striatal neurons of knockout mice (Figure 4). Sapap3 knockout in mice induces an eCB-mediated plasticity, caused by the increase in mGluR5 expression4. Thorough investigation is required to see whether these observations are related to the obsessive compulsive behavior observed in Sapap3 knockout mice4.
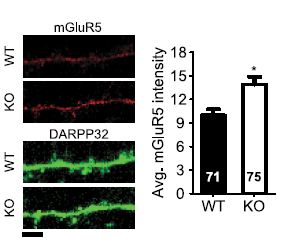
Immunocytochemical staining of wild type (WT) and Sapap3 KO (KO) mouse striatal neurons using Anti-mGluR5 (extracellular) antibody (#AGC-007). mGluR5 density is higher in Sapap3 KO striatal neurons.
Adapted from reference 4 with permission of the Society for Neuroscience.
Astrocytes were for long believed to be important for extracellular ion homeostasis and nutrient support to neurons. Ca2+ imaging studies in astrocytes have strongly suggested that these star-like shaped cells play a more prominent role in brain functions. Past studies demonstrated that mGluR5 plays an important role regarding Ca2+ signaling in astrocytes. In an elegant study, Arizono, M. et al.1 explored the detailed subcellular Ca2+ dynamics of astrocytes and the spatial regulation of mGluR5-dependent signaling in these cells. Immunocytochemical staining of rat astrocytes using Anti-mGluR5 (extracellular) antibody showed that the receptor is enriched at astrocyte processes rather than at somas, in accordance with Ca2+ imaging experiments which demonstrated that astrocytic processes are more sensitive to mGluR stimulation than the soma. The lateral movement of mGluR5 in astrocytes was monitored using quantum dot-based singleparticle tracking (QD-SPT). The procedure requires targeting streptavidinconjugated nanoparticles of semi-conductive material to the antibody after it is incubated with the cells, thereby allowing tracking the diffusion of probed mGluR5 in real-time by principles of optical relay, and eventually yielding movement trajectory and other important, often overlooked parameters1. Ultimately, QD-SPT allowed the identification of an elusive mGluR5 membrane barrier which prevents the receptor from assimilating into the plasma membrane of the soma, suggesting a close-regulation mechanism that favors controlled, specified signaling, which has important functional implications such as synaptic and vascular activity in specific subcellular domains1. In accordance with these results, disruption of the barrier resulted in disrupted Ca2+ signaling which was caused by a freely distributed mGluR5 receptor. In addition, mGluR5 localized to the soma was more static than mGluR5 in processes. Anti-P2X7 Receptor (extracellular) antibody (#APR-008) was used to show that the barrier is specific to mGluR5 since in QDSPT, P2X7 channels were able to freely diffuse between the soma and the processes. The mGluR5 specific barrier may important and crucial for local as opposed to global Ca2+signaling which could be toxic under certain disease conditions1.
Group II
mGluRs of group II include mGluR2 and mGluR3. They are negatively coupled to adenylyl cyclase and inhibit neurotransmitter release by silencing Ca2+ channels and enhancing K+ channels. They are located on presynaptic membranes where they inhibit the release of glutamate3. Agonists of this group putatively ameliorate anxiety and schizophrenic symptoms, making the receptors a favored target to treat the above disorders.
Recently, LY395756, a newly described variant of a specific agonist to this group, was shown to activate mGluR2 and to inhibit mGluR3. This selectivity was determined by using hippocampal slices from mGluR3 or mGluR2 knockout animals. Generation of mGluR3-/- rats was verified in western blot analysis using Anti-mGluR3 (extracellular) antibody (#AGC-012)3. Using a similar approach in mice, LY541850 was found to be a partial mGluR2 agonist and a full mGluR3 antagonist12. Knockout mice were generated and AntimGluR3 AntimGluR3 (extracellular) antibody was used to confirm the mGluR3 knockout12. Investigation of the mechanisms underlying mGluR II-mediated suppression of glutamate release demonstrated strong dependence of group II mGluRs on P/Q-type channel-induced AMPA EPSCs established by blocking P/Q-type voltage-gated Ca2+ channels with ω-Agatoxin IVA (#STA-500)14. Blocking N-type Ca2+ channels with ω-Conotoxin GVIA (#C-300) had no effect14.
References
- Arizono, M. et al. (2012) Sci. Signal. 5, ra27.
- Ben-Mabrouk, F. et al. (2012) Eur. J. Neurosci. 35, 1725.
- Ceolin, L. et al. (2011) J. Neurosci. 31, 6721.
- Chen, M. et al. (2011) J. Neurosci. 31, 9563.
- Ko, S.J. et al. (2012) J. Neurosci. 32, 16391.
- Krishnan, B. et al. (2011) PLoS One 6, e25639.
- Li, J.Q. et al. (2010) J. Neurochem. 112, 162.
- Meldrum, B.S. (2000) J. Nutr. 130, 1007S.
- Niswender, C.M. and Conn, P.J. (2010) Annu. Rev. Pharmacol. Toxicol. 50, 295.
- Renner, M. et al. (2010) Neuron 66, 739.
- Roselli, F. et al. (2009) PLoS One 4, e6011.
- Sanger, H. et al. (2013) Neuropharmacology 66, 264.
- Schmidt, K. et al. (2011) Eur. J. Neurosci. 34, 177.
- Wang, S. et al. (2012) Cereb. Cortex. 22, 584.
- Young, V.R. and Ajami, A.M. (2000) J. Nutr. 130, 892S.