No generalizations about the role of this enigmatic receptor in cancer can be made. As in all other aspects of its biology, the effects of p75NTR remain cell type specific, and their pattern has yet to be discovered.
p75 neurotrophin receptor (p75NTR) was the first receptor to be found for the neurotrophins (NT)1. This important family of growth receptors affect cell survival, differentiation and cell death2. Thus their connection to unchecked cell growth, i.e. cancer, is interesting and important from both a scientific and a therapeutic point of view.
The NTs (NGF, BDNF, NT3 and NT4/5) are a family of small (14 kD) basic proteins containing three cysteine bonds. All NTs are generated as pre-pro-neurotrophin precursors, about 240-260 amino acids long, which are further processed until they are secreted as mature homodimeric proteins. They bind to two types of receptors: each neurotrophin binds to a specific Trk receptor (NGF to TrkA, BDNF and NT4/5 to TrkB and NT3 to TrkC), and all neurotrophins bind to the p75NTR receptor3. The residues involved in binding of NT to Trk receptors are different than those involved in binding to p75NTR.
p75NTR is a 399 amino acid Type 1 transmembrane protein4. Its extracellular domain contains four repeated modules of six cysteines, a distinguishing structure of the tumor necrosis factor receptor (TNFR) superfamily members, of which it was the first to be discovered. The most prominent feature of the intracellular section is the death domain, an 80 amino acid module arranged in two bundles which each contain three alpha helices5. p75NTR is devoid of intrinsic catalytic activity and signal transduction necessitates the recruitment of cytoplasmic interacters.
The role of p75NTR is contradictory: p75NTR signaling has been found to cause both cell death and cell survival6. There are many factors that need to be considered: the interaction between p75NTR and Trks, the absence or presence of ligand, and the cytoplasmic interacters involved. Thus it is impossible to generalize when considering the role of p75NTR in any biological situation, and cancer is no exception.
p75NTR expression is mostly correlated with development7. In the adult, it is retained in specific cells of the central nervous system, in sympathetic and sensory neurons of the peripheral nervous system, at a number of mesenchymal/epithelial boundaries, in perivascular fibroblasts and dental pulp cells. Normal lung tissue does not express p758, however a line derived from normal breast epithelial cell9 and normal prostate10 do exhibit positive staining for p75. It is also induced by injury in a wide variety of cell types11, and with chronic diseases such as multiple sclerosis12, and Alzheimer’s.
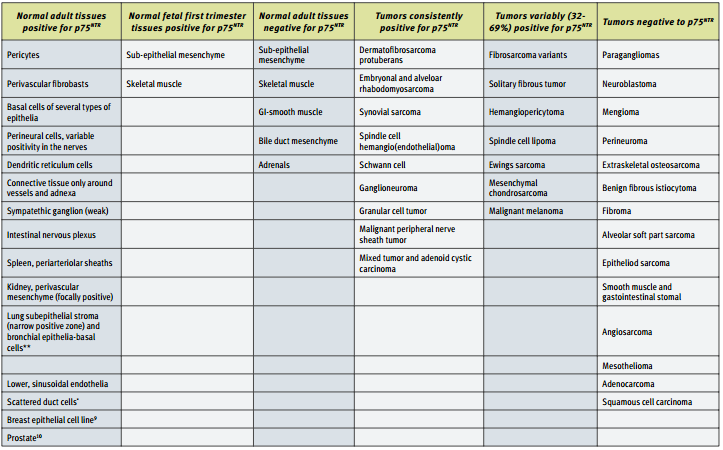
The expression patterns of p75NTR in various types of cancer, and the correlation between p75 expression and prognosis have been studied. The largest study done involved 1,150 tumors and fetal and adult normal tissue14. A summary of these results can be found in the table below. The striking observation to be made from these results is that p75NTR seems to be absent in as many cancers as it is present. Thus, the expression of p75NTR is not an indication of the cancerous phenotype. However, p75NTR can be used as a marker in certain specific situations, such as to distinguish DFSP from benign fibrous histiocytoma14.
In prostate carcinoma p75NTR has been found to act as a tumor suppressor10. The expression of p75NTR in tissue sections from radical prostatectomies and in the prostate tumor cell lines DU-145, PC-3, LNCaP and TSU-pr1 (established from metastasis) was examined. It was found that there was a progressive loss of p75NTRexpression in prostate epithelial cells during the malignant progression of organ confined adenocarcinomas with complete loss of expression in the naturally occurring prostate tumor cell lines.
These researchers also investigated proliferation in the human T24 bladder cell line, (which does not express endogenous p75NTR), transfected with varying levels of p75NTR. They found p75NTR to be an effective tumor suppressor of bladder cancer cell growth in vivo15. This was attributed to the alteration of cell cycle regulator proteins by p75NTR and was ameliorated by NGF. They also found that p75NTR protein expression induced a dose-dependent increase in pro-apoptotic effectors and a decrease in prosurvival effectors of mitochondrial-mediated apoptosis16.
In one of the first studies on the effects of NT on breast cancer, it was found that NGF could stimulate the proliferation of breast cancer cell lines MCF-7 and MDA-MB231 but was unable to stimulate growth of normal epithelial breast cells in culture9. Both the cancerous and the normal cells expressed p75NTR and TrkA. In addition, breast cancer cells, but not normal breast cells, produce NGF17. Further studies showed that the mitogenic effect of NGF required TrkA, but the anti-apoptotic effect was mediated by p75NTR through the NF-KB pathway18. The TNF-receptor associated death domain protein (TRADD) interacts with p75NTR upon NGF stimulation, thus mediating the anti-apoptotic activity of NGF in breast cancer cells19. The level of p75NTR has been shown to be related to tumor type, with low levels being more frequently found in ductular type tumors. In an immunohistochemical study of human invasive ductal breast carcinoma it was found that patients who were NGF positive and p75NTR negative had significantly poorer survival rate than those who were NGF negative and p75NTR positive20. The phenotypic alterations along the progression of breast carcinoma from primary tumor to pleural effusion has shown that p75NTR expression was less frequent in effusions compared to both primary tumor and lymph node metastases21. The loss of the p75NTR related pro-apoptotic mechanism may provide one biological explanation as to why the presence of malignant cells in effusions is associated with a rapidly fatal course in both breast cancer and malignant mesothelioma21.
The connection between neurotrophins, neurotrophin receptors and the ability of cancer cells to metastasize and colonize tissues has been investigated. This process is especially important in the case of malignant melanoma, which has one of the highest frequencies of metastasis to the brain22. It was known that the overexpression of p75NTR is frequently associated with advanced stages of human melanoma progression23. The human melanoma MeWo cell line and its sublines 70W and 3S5 express varying levels of p75NTR, the highest being found in the 70W subline24. The expression level of p75NTR was to correlate with a number of properties:
- The ability of the cells to metastasize in nude mice. The A875 cell line, which also expresses high levels of p75NTR, is also highly metastatic.
- The extent of invasion of extracellular matrix stimulated by NGF.
- The ability of the cells to grow under stress conditions (normal melanocytes did not grow under these conditions).
It was hypothesized that brain metastasis essentially represents a traumatic event related to brain injury processes. Thus the same upregulation of NT and NT receptors found in response to chemical or mechanical brain injury is also found when the brain is colonized by metastatic melanoma cells25.
The effects of p75NTR on neuroblastoma cells were intriguing. SK-N-BE neuroblastoma cell clones transfected with p75NTR and lacking TrkA undergo extensive spontaneous apoptosis from which they are rescued by NGF26. This process was postulated to involve nitric oxide synthesase.27 Another study also confirmed this effect of NGF, but found that it was reversed in another cell line, Neuro-2a, which also expresses p75NTR but not TrkA28. The authors note that the level of expression of p75NTR was different in the two lines, being higher in Neuro-2a and speculate that this may explain their conflicting results, however they caution that many other factors, most importantly the distribution of the p75NTR (in domains such as microlipid rafts) need to be determined before jumping to any conclusions.

A. Distribution of the neurotophin receptor p75 using Mouse Anti-Rat p75 NGF Receptor (extracellular) Antibody(#AN-170) in the diagonal band of the rat basal forebrain shown by indirect immunofluorescence labelling. B. Exclusive co-expression of p75 (yellowish dots) in cholinergic projection neurons revealed by concomitant carbocyanine staining of choline acetyltransferase (ChAT, green).
Picture obtained by a Zeiss LSM 510 Meta, from Wolfgang Hartig & Jens Grosche, PFI, Leipzig Univ., Germany).

Expression of p75NTR in normal rat tissue (right blot) and in human cancer cell lines (left blot). p75NTR was visualized with Anti-p75 NGF Receptor (extracellular) Antibody (#ANT-007) diluted 1:200. Note that human cancer cell lines from hemapoietic origin show high p75NTR expression, while cell lines from prostate and colon cancer origin show lower levels. Interestingly, p75NTR from rat (right blot and the C6 cell line) and human (left blot) samples run with a different apparent MW, probably due to species-specific differential glycosylation.
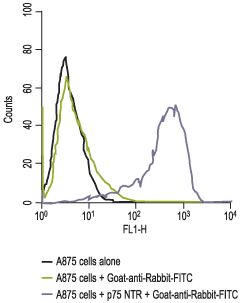

Staining of human melanoma cells A875 with Anti-p75 NGF Receptor (extracellular) Antibody (#ANT-007). Cells were incubated with Anti-p75 NGF Receptor (extracellular) Antibody (I:200) (A and B) or with Anti-p75 NGF Receptor (extracellular) Antibody preincubated with the negative control antigen (C and D), followed by goat anti-rabbit-Alexa Fluor 555 (Red), (x100).
References
- Rodriguez-Tebar, A. et al. (1992) EMBO J. 11, 917.
- Barker, P.A. (2004) Neuron 42, 529.
- Klein, R. et al. (1991) Cell 66, 395.
- Large, T.H. et al. (1989) Neuron 2, 1123.
- Roux, P. et al. (2002) Prog. Neurobiol. 67, 203.
- Bronfman, F.C. and Fainzilber, M. (2004) EMBO Reports 5, 867.
- Barker, P.A. (1998) Cell Death Differ. 5, 346.
- Ricci, A. et al. (2001) Am. J. Respir. Cell Mol. Biol. 25, 439.
- Descamps, S. et al. (1998) J. Biol. Chem. 273, 16659.
- Krygier, S. and Djakiew, D. (2001) Mol.Carcinog. 31, 46.
- Hempstead, B. (2002) Curr.Opin.Neurobiol. 12, 260.
- Copray, S. (2004) J. Neuroimmunol. 148, 41.
- Correia, L. et al. (1998) Brain Res. 809, 288.
- Fanburg-Smith, F. C. (2001) Hum. Pathol. 32, 976.
- Khwaja, F. and Djakiew, D. (2003) Mol.Carcinog 36, 153.
- Tabassum, A. et al. (2003) Int. J. Cancer 105, 47.
- Dolle, L. et al. (2004) Curr. Cancer Drug Targets 4, 463.
- Descamps, S. et al. (2001) J. Biol. Chem. 276, 17864.
- Descamps, S. et al. (2003) J. Biol. Chem. 278, 16952.
- Sakamoto, Y. et al. (2001) Oncol. Rep. 8, 973.
- Davidson, B. et al. (2004) Breast Cancer Res. Treat. 83, 119.
- Denkins, Y. et al. (2004) Neuro-Oncology 6, 154.
- Hermann, J. L. et al. (1993) Mol. Biol. Cell 4, 1205.
- Marchetti, D. et al. (2004) J. Cell. Biochem. 91, 206.
- Marchetti, D. et al (2003) Pathol. Oncol. Res. 9, 147.
- Bunone, G. et al. (1997) Oncogene, 14, 1463.
- Lievremont, J. P. et al. (1999) J. Biol. Chem. 274, 15466.
- Evangelopoulos, M.E. et al. (2004) J. Neuro-Oncol. 66, 101.