Venomous creatures have a sophisticated mechanism for prey capture which includes a vast array of biologically active compounds, such as enzymes, proteins, peptides and small molecular weight compounds. These substances target an immense number of receptors and membrane proteins with high affinity, selectivity and potency, and can serve as potential drugs or scaffolds for drug design.
Introduction
A large number of organisms produce and secrete venoms to defend themselves and to capture prey. Venom is a rich source of biochemically active enzymes, proteins, peptides and low molecular weight substances. Toxins isolated from the venom either inhibit or activate a vast number of targets such as ion channels, acetylcholine receptors, acetylcholinesterase, membranes, coagulant/anticoagulant pathways, and metalloproteases, with high selectivity and affinity. They can be roughly divided into non-peptide and peptide toxins. Non-peptide toxins have been isolated from algae, plants, dinoflagellate, fish and from higher organisms which accumulate alkaloids through their diet as exemplified in toxic frogs. Peptide toxins are generally synthesized in the venomous ducts of poisonous creatures. The majority of the data acquired to date has been from toxins isolated from the venoms of snakes, scorpions, spiders, marine snails (Conus genus) and sea anemones. Toxins isolated from venomous animals are usually small, ranging from 8-70 amino acids, with relatively small scaffold structures, which are highly compact and stabilized by either disulfide bonds or by hydrogen bonds made from post-translational-modified amino acids. In a number of toxins, the active residues responsible for the toxin activity have been identified, thus enabling the rational design of small molecular weight compounds or peptomimetics.
The pharmaceutical industry has recognized the enormous potential inherent in these venom peptides and has begun to exploit the selectivity and sensitivity fine tuned by evolution. This review will focus on peptides and toxin mimetics that are currently being evaluated as possible drugs for the treatment of pain, epilepsy, cardiovascular disorders, cancer and other neurological disorders (See Table), (for recent and comprehensive reviews please refer to references 1-10).
Pain
Recurrent pain was recently termed the “silent epidemic”, since one out of six people in the Western World suffers from pain, costing the American public alone approximately $100 billion each year in health care, compensation, and litigation11,12. Chronic pain is associated with conditions such as back injury, migraine headaches, arthritis, herpes zoster, diabetic neuropathy, temporomandibular joint syndrome, and cancer. Effective treatment options are limited to opioids (morphine and related drugs) and non-steroidal anti-inflammatories (NSAIDs), yet opioids have significant potential side effects, and NSAIDs are ineffective for moderate-to-severe pain. Pain can be generated by nociceptors stimulated by thermal, mechanical, chemical or inflammatory stimuli, and a pivotal role for ion channels has been established13 (please refer to the article “Contribution of Ion Channels in Pain Sensation” Modulator 19, 2005 issue). Pain signals can be blocked at a number of sites along the pain pathway (See Figure). The large list of neurotransmitters and receptors identified along the pain pathway indicate that there may be many therapeutic possibilities for the pharmacological control of the transmission of nociceptive information to the brain.
Voltage-Gated Ca2+ Channels (VGCC)
Cav2.2 (α1B, N-type) was shown to control transmission at CNS and PNS synapses including in the transmission of pain signals at the spinal level. A PNS-specific CaV2.2 splice variant is highly expressed in the superficial layer of the dorsal horn, which is considered to be responsible for the nociceptive pathway of the spinal cord14. CaV2.2 was found to be up-regulated in the spinal cord during chronic pain states, along with the auxiliary α2δ-1 subunit15,16. Blockers of N-type channels were shown to block Ca2+ influx and thus the release of substance P in the spinal cord17. Furthermore, N-type channels are susceptible to modulation by μ-opioid peptide receptor agonists, such as morphine18. Recently it was proposed that T-type and possibly P/Q-Type Ca2+ channels may participate in pain pathways, and may serve as possible therapeutic targets14,19,20.
A large array of VGCC peptide inhibitors were isolated from the venoms of cone snails, spiders and snakes. The most selective inhibitors of N- type Ca2+ channels known to date were isolated from Conus geographus (ω-Conotoxin GVIA (#C-300)), Conus magus (ω-Conotoxin MVIIA (#C-670)), and Conus catus, (ω-Conotoxin CVID). ω-Conotoxin MVIIA, a 25 amino acid peptide is a highly potent and selective blocker of N-type VGCC21. A synthetic ω-Conotoxin MVIIA analog, Prialt™ (previously called SNX-111 and Ziconotide) initially developed by Neurex Corporation and currently being commercialized by Elan Corporation for the treatment of severe chronic inflammatory and neuropathic pain associated with cancer and AIDS22,23. Although injected intrathecally, it was shown that Prialt™ is about 1000-fold more potent then morphine, lacking the tolerance or addiction usually associated with opiates24. However, Prialt™ administration results in severe side effects, including hypotension, sedation and confusion25. During December 2004, Elan Corporation was granted FDA approval for Prialt™ (ziconotide intrathecal infusion) for the management of severe chronic pain. ω-Conotoxin CVID analog, AM336, which is being developed by Amrad Corporation, has been in Phase II clinical trial since 2002 with cancer patients suffering severe chronic pain. AM336 was shown to inhibit a PNS-specific splice variant N-type VGCC associated with transmitter release from preganglionic nerve terminal and displays a wider therapeutic index than ω-Conotoxin MVIIA17,26. Recently, attempts were made to “convert” the toxin’s pivotal binding residues into smaller molecular weight compounds, which might have superior pharmacological qualities in terms of formulation, production, metabolic stability and delivery27-31. One approach compared the structure of ω-Conotoxin CVID against virtual screening libraries resulting in a cyclic peptide with D-amino acids having an IC50 of ~20 μM and apparent selectivity between N- and P/Q- type VGCC27. Furthermore, using alkylphenyl ether based analogues which mimic three key amino acids of the toxins (Arg-10, Leu-11 and Tyr-13), leads to three compounds which have an apparent IC50 of ~3 μM28. These and other experiments show that it is possible to design smaller entities which are active and selective.
Voltage-Gated Na+ Channels (VGSC)
VGSC modulators have been isolated from the venom of a variety of organisms, including spiders, sea anemones, scorpions, and cone snails1,8. There is a great need for specific venom peptides that can discriminate between the different VGSCs. A possibly abundant source for VGSC modulators was found in the venoms of cone snail venoms. Two main classes of conotoxins that affect VGSC are the μ- and δ-Conotoxins3,8. Recently the μ-Conotoxin SmIIIA toxin was isolated from the venom of Conus stercusmuscarum, which has been shown to block TTX-R currents in amphibian sympathetic and sensory neurons32,33. The authors speculate that the irreversible current block in the frog DRG was by inhibition of NaV1.8 and NaV1.9 channels, while the reversible inhibition of frog skeletal muscle is due to NaV1.4 channel inhibition32. μ-Conotoxin SmIIIA or its mimetics may be an attractive toxin for pain treatment.
Neuronal Nicotinic Acetylcholine Receptors (nAChRs)
Nicotinic acetylcholine receptors (nAChRs) are a family of ligand-gated cation channels whose endogenous ligand is acetylcholine (ACh) and which are activated by the alkaloid, nicotine. These pentameric conductance channels for Ca2+, K+, and Na+ are formed from a number of homologous subunits, and various combinations of these subunits result in channels which differ in their pharmacology and tissue distribution34. A class of conotoxins block nAChR and can differentiate between neuronal, skeletal muscle and sub-population isoforms of receptors, termed α- and αΑ- Conotoxins3, 8, 35. The latter toxins are termed alpha toxins as compared to a similarly acting α-Bungarotoxin (#B-100), a blocker of muscle-type nAChR. Attempts to understand the mechanism of the selectivity have been reported36, 37. The α-Conotoxins inhibit the signal transmission at the postsynaptic junction (neuronal or neuromuscular), by binding to the α-subunits of the nAChR, thus blocking the binding of acetylcholine and other agonists, which in turn inhibits the influx of Na+ required for action potential propagation.
α-Conotoxin Vc1.1 (#STV-500) (also called ACV1), is a 16 amino acid peptide, containing two disulfide bonds. Its sequence was deduced from the nucleotide sequence of the conopeptide gene cDNA amplified from mRNA extracted from the venom duct of Conus victoriae. Vc1.1 was active as an antagonist of neuronal nAChRs in receptor binding and functional studies on bovine chromaffin cells. It also suppressed the vascular responses to C-fiber activation, and accelerated the functional recovery of injured peripheral nerves in rats. These peripheral unmyelinated sensory nerves are involved in pain transmission38. Recently, Metabolic Pharmaceuticals Ltd., who are developing ACV1 as an analgesic for the treatment of chronic neuropathic pain, has begun preclinical and formal safety trials with this synthetic conopeptide. According to Metabolic Pharmaceuticals, preclinical trials show almost full relief of chronic pain with no apparent side effects and Phase I clinical trials are set to begin in early 2005.
Noradrenaline Transporter Inhibitors
Noradrenaline (or norepinephrine) (NE) participates in a number of biological pathways including the regulation of mood and sleep, expression of behavior, alertness and arousal. In episodes of pain, the elevation of NE levels in the spinal cord results in an inhibition of pain messages. The norepinephrine transporter (NET) returns noradrenaline to the synapses.
Among the A-super-family of conotoxins, the ρ- and χ-Conotoxins are known to modulate the α1-adrenorecetor and neuronal noradrenaline transporter, exemplified by the two Conotoxins ρTIA and χMrlA isolated from Conus tulipa and Conus marmoreus, respectively38,39,40.
The synthetic χMrlA, termed Xen2174, acts by selectivity binding to NET and abolishing its ability to transport NE from the synapse back into the nerve ending. Xen2174, developed by Xenome Ltd., has recently entered Phase I clinical trials (intrathecal) for relieving nociceptive and neuropathic pain, while ρTIA is currently in preclinical trials3. Preclinical trials of Xen2174 in experimental animal pain models show no side effects and a high therapeutic index. Both ρTIA and χMrlA lack the common and often therapeutically limiting pharmacology of α1– adrenoceptor antagonists (α2-adrenoceptor and Na+ channel inhibition) and NET inhibitors (α1-adrenoceptor and muscarinic ACh receptor antagonism), and thus may be useful clinically39,40.
Neurotensin Receptor Agonist
Contulakin-G (#GPC-100) is a novel 16-amino acid conopeptide originally isolated from the venom of the marine snail Conus geographus41. Cognetix Inc. is developing Contulakin-G (CGX-1160) for the short-term management of post-operative pain. CGX-1160 interacts with the neurotensin receptor 1 with 100-fold less potency (than neurotensin), but is 100-fold more potent as an analgesic, suggesting additional modes of action besides NT binding1. CGX-1160 has completed early Phase I safety studies in humans and has demonstrated efficacy in a broad range of preclinical models of acute and chronic pain.
NMDA Receptor Antagonist
Glutamate is the major excitatory neurotransmitter in the mammalian CNS. Upon release from presynaptic terminals, glutamate binds to postsynaptic ionotrophic receptors NMDA, kainite and AMPA. Glutamate acting on NMDA receptors is responsible for the initiation of CNS sensitization and hyperexcitability of spinal cord neurons upon nerve injury42,43. Non-specific NMDA antagonists relieve injury–induced pain, but have pronounced side effects43,44,45,50.
Conantokins were identified as a group of peptides that competitively inhibit glutamate activation, especially through NR2B or NR2B and NR2A subunits of NMDA receptors, and can discriminate between the different NMDA receptor types in the human brain7,8,43,46. Conantokin-G (#STC-666), a 17-amino acid peptide isolated from Conus geographus selectively inhibits NR2B, while its related isoform, Conantokin-T, 21 amino acids long, isolated from Conus tulipa, inhibits both NR2B and NR2A receptors8,47. Both Con-G and Con-T lack disulfide bonds, and their structural stability is due to five post-translationally modifi
ed residues of the nonstandard amino acid γ-carboxyglutamate (Gla)48,49. Cognetix Inc. is currently developing conantokin-G synthetic derivative (CGX-1007) as an anti-nociceptive drug and for control of seizures in intractable epilepsy, and is currently in Phase II clinical trials43.
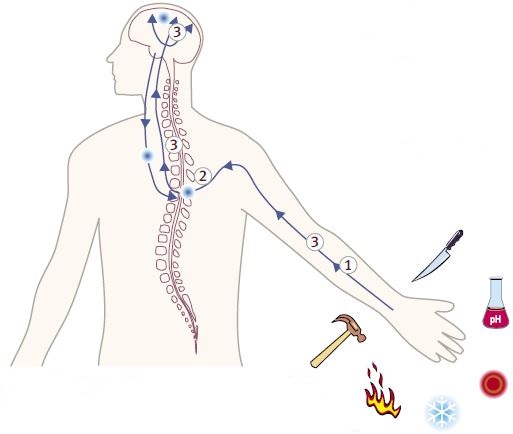
Pain can be generated by nociceptors from a number of stimuli including, heat, cold, mechanosensation, inflammation, injury and pH. Venom toxins intersect pain signaling by inhibiting or stimulating channels and receptors along the pathway. The possible localization of the pain pathway targets are illustrated above.
1- Voltage-Gated Na+ Channels.2- Voltage-Gated Ca2+ Channels.
3- Neuronal Nicotinic Acetylcholine Receptors Noradrenaline Transporter Neurotensin Receptor Agonist NMDA Receptor Antagonist.
Epilepsy
According to the Epilepsy Foundation of America, an estimated 1% of the total population suffers from epilepsy and seizures, afflicting more than 2.3 million Americans, with combined direct and indirect costs to the American economy of $12.5 billion. Total market volume of anti-epileptic drugs reaches $1.9 billion a year worldwide, with a 5% annual growth rate.
NMDA Receptor Antagonist
NMDA receptors have been shown to participate in a number of CNS malfunctions. CGX-1007 (#STC-666) (see above) is currently in Phase II clinical trials as an anticonvulsant and for intractable epilepsy (when delivered directly into the central nervous system). The Phase I, randomized, double blind, placebo-controlled trial involved intravenous delivery of single, escalating doses of CGX-1007 in healthy, normal subjects to determine safety of the compound when administered to the systemic circulation. The results of the Phase I trial demonstrated that CGX-1007 was safe, with no clinically remarkable drug-related adverse experiences observed. Recently it was shown that although considered NR2B-specific, CGX- 1007 is less specific or acts differently, than the investigational CI-1041 compound, in corneal kindled rats and in an NMDA receptor mediated excitatory postsynaptic currents model (N-EPSC)44,45.

Stroke, Neuroprotection and Cardiovascular Disorders
Stroke and myocardial ischemia affect more than 65 million people in the United States and are the leading cause of death. Stroke is the leading cause of adult disability. Each year more than 700,000 Americans suffer a stroke, and one in five of them die. The total market volume of cardiovascular disorders drugs is estimated to be around $60 billion a year worldwide51.
Angiotensin Converting Enzyme (ACE)
ACE is an essential enzyme required for production of angiotensin, associated with hypertension due to vasoconstriction. Anti- hypertensive effect is achieved by inhibiting ACE. Capoten® (captopril) is a small molecular mimetic compound derived from a toxin found in the venom of the Brazilian arrowhead viper (Bothrops jaracusa) developed by Bristol-Myers Squibb52.
Platelet Aggregation and Blood Clotting Inhibitors (inhibitors of platelet glycoprotein IIb/IIIa receptor)
Schering-Plough and Millennium Pharmaceuticals have been granted FDA approval for Integrilin® (Eptifibatide), a synthetic analog of barbourin, for the treatment of severe cardiovascular diseases, namely, anticoagulation in patients with acute coronary syndrome (ACS) and for patients without ACS undergoing percutaneous coronary intervention (angioplasty). Integrilin® is a heptapeptide derived from a protein found in the venom of the southeastern pygmy rattlesnake (Sistrurus miliarius barbouri)53. Integrilin® acts as a parenteral platelet receptor glycoprotein IIb IIIa (GPIIb-IIIa) inhibitor and blocks platelet aggregation, a crucial event in thrombosis.
Aggrastat® (Tirofiban), developed by Merck, is also a GPIIb-IIIa inhibitor, however this drug was modeled on the structure of Echistatin, a derivative of the anticoagulant found in the venom of the African saw-scaled viper (Echis carinatus). Aggrastat® was the first GPIIb-IIIa inhibitor to be launched, but it is only approved for use with heparin and aspirin for the treatment of ACS54.
ViprinexTM (Ancrod), a compound isolated from the venom of the Malaysian pit viper (Agkistrodon rhodostoma) is in late Phase III trials by Neurobiological Industries Inc. for use in the treatment of heparin-induced thrombocytopenia (deficit of platelets). Researchers found that blood failed to clot in animals bitten by these snakes. ViprinexTM removes fibrinogen from the blood, improving blood flow, a useful property that also has potential for the treatment of stroke.
Fibrin Thrombolytic Agent
Nuvelo Inc. is currently evaluating Alfimeprase, a synthetic version of fibrolase, a protein isolated from the venom of the southern copperhead viper (Agkistrodon contortrix), as an anticoagulant for the treatment of ischemic stroke and catheter occlusion. Alfimeprase, now in Phase II, was shown to directly degrade fibrin, producing a rapid dissolution of blood clots. Furthermore, Alfimeprase is currently in Phase II for catheter occlusion.
Thrombin Inhibitors
AstraZeneca is seeking FDA approval for ExantaTM (Ximelagatran) for treatment of patients with atrial fibrillation and patients at risk for blood clots. The orally active thrombin inhibitor was designed based on a cobra venom peptide. It is already on sale in Europe as a treatment to prevent blood clotting after orthopedic surgery55.
NMDA Blockers
Delucemine (NPS 1506) is a compound being developed by NPS Pharmaceuticals as a means of protecting brain cells in ischaemia victims. Its structure was based on a spider venom toxin56,57. Phase I clinical trials with delucemine are currently underway in patients suffering from stroke and acute depression. NPS 1506 blocks NMDA receptors on neurons, thus preventing excessive Ca2+ influx during ischaemia.

Cancer
Current estimates by the American Cancer Society indicate that approximately 1.3 million individuals in the U.S. were diagnosed with cancer and there were about 500,000 cancer related deaths in 2002. Approximately $10 billion is spent on cancer drugs annually and cancer drug expenditures account for roughly 8% of total U.S. drug sales.
Cl– Channels
Chlorotoxin (#RTC-450 [recombinant] or #STC-460 [synthetic]) is a 36-amino acid peptide that was originally isolated from the venom of the Leiurus quinquestriatus scorpion as a putative Cl– channel inhibitor58. It was later found that Chlorotoxin could inhibit invasiveness of glioma cells in vitro. This inhibition was attributed to the ability of Chlorotoxin to block an unidentified Cl– channel that was putatively involved in the process of regulatory volume decrease, a key step in cell migration. Interestingly, Chlorotoxin was found to bind specifically to glioma cell lines and primary cultures, but not to normal brain cells59,60. Positive staining towards the labeled Chlorotoxin was found in other solid tumor cell lines, including non-small cell lung carcinoma, breast, prostate, melanoma, and colon cancers. Recent studies have shown that contrary to the original hypothesis, the specific target of Chlorotoxin on the surface of glioma cells might be matrix metalloproteinase-2 (MMP-2) protein and not a Cl– channel61. Recently TransMolecular Inc. has initiated Phase II clinical trials with its iodinated Chlorotoxin derivate TM-601 (131I-Chlorotoxin) for the treatment of brain tumors. TM-701, a derivative of TM-601, shares the same mechanism of action, but is used without a radioisotope. TM- 701 is being developed as a chronic monotherapy and pharmaceutical sensitizer when administered with commonly used drug cocktails for treating cancer and is currently in preclinical trials. Other derivatives of TM-601 are being tested for use in in vivo imaging and diagnostic test kits.
Integrins
Integrins are a family of cell surface proteins, found on many cell types that mediate interactions between cells, and between cells and their surroundings. Specific integrin isoforms are upregulated during tumor growth71. Contortrostatin, a protein extracted from the venom of the southern copperhead viper (Agkistrodon contortrix), binds to integrins on the surface of cancer cells and inhibits tumor growth and metastasis, while no cytotoxic effect on human breast cancer cells is observed62. PB2 (Contortrostatin), is cytostatic, ‘freezing’ tumor cells rather than killing them63. It is presently in preclinical research for the treatment of breast cancer by Pivotal Biosciences and the University of Southern California.

Diabetes
Glucagon-Like Peptide-1 (GLP-1)
Glucagon-Like Peptide-1 is an insulinotopic hormone secreted from endocrine cells of the small and large intestine in a nutrient-dependent manner. GLP-1 stimulates insulin secretion and modulates gastric emptying to slow the entry of ingested sugars into the bloodstream. The GLP-1 related peptide is a peptide initially derived from the salivary secretions of the Gila monster (Heloderma suspectum), a large venomous lizard. Amylin Pharmaceuticals is developing a synthetic version of Exenatide (synthetic exendin-4), a 39-amino acid peptide, currently in Pre-Phase III for use in treating type-2 diabetes and related metabolic disorders. Diabetic animal models have demonstrated that Exenatide is biologically active when administered via oral, sublingual, pulmonary, tracheal and nasal routes. Furthermore, GLP-1 like peptides share structural homology to α-Latrotoxin (#LSP-130), isolated from the venom of the black widow spider and might have potential in the treatment of Alzheimer’s disease64,65.
Immunosuppressants and Autoimmune Disorders
The existence and participation of the voltage-dependent K+ channel KV1.3 and the Ca2+-activated intermediate K+ channel IKCa1 (KCa3.1) in T-lymphocyte activation is well established1,9. Furthermore, a marked elevation of KV1.3 is reported in encephalitogenic T-cells, which mediate demyelination of axons in the brain and spinal cord, the hallmark of multiple sclerosis66. The use of specific blockers for KV channels might have therapeutic potential for treatment of autoimmune disease, and as immunosuppressants for transplantations. In vitro studies, the use of peptidyl toxins has indicated that blockage of KV1.3 inhibits T-cell activation, suggesting that KV1.3 may be a target for immunosuppression9. This concept was verified by in vivo experiments on peripheral T-cells of mini-swine using Margatoxin (#STM-325) as specific KV1.3 toxin67,68. Side effects of Margatoxin administration have been observed, mainly in the enteric nervous system which is expected for all non-specific KV1.3 toxins1,9. Furthermore, high serum concentrations of Margatoxin caused transient hyperactivity in pigs, indicating possible effects on KV1.1 and KV1.2 channels in the brain. Stichodactyla toxin (ShK) (#STS-400, #STS-390 [Dalazatide, ShK-186 [SL5]], #STS-400-AR [ATTO-590], #STS-400-B [Biotinylated]), a toxin isolated form the venom of sea anemone Stichdactyla helianthus, has relatively similar affinities towards KV1.3 and KV1.169,72. A ShK mutant, ShK-Dap22 co-administered with TRAM-34, inhibitor of IKCa1, was tested in experimental autoimmune encephalomyelitis (EAE) animals (a model for multiple sclerosis), and was shown to effectively prevent lethal EAE9,66.
Conclusion
It is estimated that between 500 to 700 conus species exist, each possessing between 50-200 conopeptides in their venom. Thus, theoretically, over 50,000 pharmacologically active components can be found in the conus genus venoms, whereas only a small fraction has been studied to date8. Furthermore, the exact venom composition of other venomous creatures are planned to be sequenced in the Venom Genome project70. The precise protein-protein interactions between the venom peptides and the different channels might enlighten the possible selectivity issues that, in turn, might enable the design of more potent and selective toxins and of new small molecules with higher selectivities for new and safer drugs. Taken together, it seems that nature has evolved the venoms into a huge pharmacological library of active pharmaceuticals with high selectivities and affinities, which could be explored as therapeutics or serve as a template for drug design. The mechanism of action of each toxin family is different, thus each needs to be evaluated for its therapeutic potential. In conclusion, the large number of venom components may possibly serve as drug libraries, diagnostic tools, and for target specific research tools.
Acknowledgments
I would like to thank Dr. Maria L. Garcia, Merck Research Laboratories, Rahway, N.J., USA and Dr. Bruce G. Livett, University of Melbourne, Parkville, Victoria, Australia, for their help and critical reviews of this manuscript.
References
- Lewis, R.J. and Garcia, M.L. (2003) Nat. Rev. Drug. Discov. 2, 790.
- Aneiros, A. and Garateix, A. (2004) J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 803, 41.
- Livett, B.G. et al. (2004) Curr. Med. Chem. 11, 1715.
- Alonso, D. et al. (2003) Mini. Rev. Med. Chem. 3, 785.
- Rajendra, W. et al. (2004) Toxicon. 44, 1.
- Rajendra, W. et al. (2004) Brain. Res. Brain. Res. Rev. 45, 125.
- Shen G.S. et al. (2000) Drug Discov. Today. 5, 98.
- Terlau, H. and Olivera, B.M. (2004) Physiol. Rev. 84, 41.
- Vianna-Jorge, R. and Suarez-Kurtz, G. (2004) Biodrugs 18, 329.
- Ault, A. (2004) The Scientist. 19, 43.
- Loeser, J. D. et al. (2001) Bonica’s Management of Pain (Lippincott, Philadelphia).
- Palmer, K.T. et al. (2000) B.M.J. 320, 1577.
- Julius, D. and Basbaum, A.I. (2001) Nature. 413, 203.
- Altier, C. and Zamponi, G.W. (2004) Trends Pharmacol. Sci. 25, 465.
- Luo, Z.D. et al. (2001) J. Neurosci. 21, 1868.
- Cizkova, D. et al. (2001) Exp. Brain. Res. 147, 456.
- Smith, M.T. et al. (2002) Pain 96, 119.
- Connor, M. et al. (1999) Br.J. Pharmacol. 128, 1561.
- Matthews, E.A. and Dickenson, A.H. (2001), Pain. 92, 235.
- Fukuizumi, T. et al. (2003) Life Sci. 73, 2873.
- Olivera, B.M. et al. (1987) Biochemistry. 26, 2086.
- Staats, P.S. et al. (2004) J. Am. Med. Assoc. 291, 63.
- Wermeling, D. et al. (2003) J. Clin. Pharmacol. 43, 624.
- Scott, D.A. et al. (2002) Eur. J. Pharmacol. 451, 279.
- Penn, R.D. and Paice, J.A. (2000) Pain. 85, 291.
- Adams, D.J. et al. (2003) J. Biol. Chem. 278, 4057.
- Schroeder, C.I. et al. (2004) Mol. Devers. 8, 127.
- Menzler, S. et al. (2000) Bioorg. Med. Chem. Lett. 10, 345.
- Flinn, J. P. et al. (1999) Eur. J. Biochem. 262, 447.
- Baell, J. B. et al. (2002) J. Comput. Aided Mol. Des. 15, 1119.
- Pallaghy, P.K. and Norton, R.S. (2000) Biopolymers. 54, 173.
- West, P.J. et al. (2002) Biochemistry. 41, 15388.
- Keizer, D.W. et al. (2003) J. Biol. Chem. 278, 46805.
- Lloyd, G.K. and Williams, M. (2000) J. Pharmacol. Exp. Ther. 292, 461.
- Nicke, A. et al. (2004) Eur. J. Biochem. 271, 2305.
- Millard, E.L. et al. (2004) Eur. J. Biochem. 271, 2320.
- Dutertre, S. and Lewis R.J. (2004) Eur. J. Biochem. 271, 2327.
- Sandall, D.W. et al. (2003) Biochemistry. 42, 6904.
- Sharpe, I.A. et al. (2003) J. Biol. Chem. 278, 40317.
- Sharpe, I.A. et al. (2001) Nat. Neurosci. 4, 902.
- Craig, A.G. et al. (1999) J. Biol. Chem. 274, 13752.
- Sotgiu, M.L. and Biella, G. (2000) Neurosci. Lett. 283, 153.
- Malmberg, A.B. et al. (2003) Pain. 101, 109.
- Barton, M.E. et al. (2004) Epilepsy Res. 59, 13.
- Barton, M.E. and While, H.S. (2004) Epilepsy Res. 59, 1.
- Ragnarsson, L. et al. (2000) J. Neurochem. 81, 765.
- Klein, R.C. et al. (2001) J. Biol. Chem. 276, 26860.
- Rigby, A.C. et al. (1997) Biochemistry. 36, 6906.
- Skjaebaek, N. et al. (1997) J. Biol. Chem. 272, 2291.
- LoGrasso, P. and McKelvy, J. (2003) Curr. Opin. Chem. Biol. 7, 452.
- http://www.innovations-report.de.
- Cushman, D.W. and Ondetti, M.A. (1999) Nat. Med. 5, 1110.
- Rossi, M.L. and Zavalloni, D. (2004) Mini. Rev. Med. Chem. 4, 703.
- Bennett, J.S. (2001) Annu. Rev. Med. 52, 161.
- Steinmetzer, T. and Sturzebecher, J. (2004) Curr. Med. Chem. 11, 2297.
- Mueller, A.L. et al. (1999) Ann. N. Y. Acad. Sci. 890, 450.
- Lorber, A. et al. (2000) J. Neurosurg. Anesthesiol. 12, 345.
- DeBin, J.A. et al. (1991) Toxicon. 29, 1403.
- Soroceanu, L. et al. (1999) J. Neurosci. 19, 5942.
- Soroceanu, L. et al. (1998) Cancer Res. 58, 4871.
- Deshane, J. et al. (2003) J. Biol. Chem. 278, 4135.
- Swenson, S. et al. (2004) Mol. Cancer Ther. 3, 499.
- Zhou, Q. et al. (2000) Breast Cancer Res. Treat. 61, 249.
- Holz, G.G. and Habener, J.F. (1998) Comp. Biochem. Physiol. B. 121, 177.
- Perry, T. and Greig, N.H. (2002) J. Alzheimers Dis. 4, 487.
- Beeton, C. et al. (2001) Proc. Natl. Acad. Sci. USA. 98, 13942.
- Koo, G.C. et al. (1997) J. Immunol. 158, 5120.
- Shah, K. et al. (2003) Cell. Immunol. 221, 100.
- Kalman, K. et al. (1998) J. Biol. Chem. 273, 32697.
- http://www.toxinology.org/.
- Guo, W. and Giancotti, F.G. (2004) Nat. Rev. Mol. Cell. Biol. 10, 816.
- Middleton, R.E. et al. (2003) Biochemistry. 42, 13698.