In many physiological systems, voltage dependent Ca2+ (CaV) channels are the molecular link between cellular membrane potential and intracellular Ca2+ signaling. As such, CaV channels are drug targets for several cardiovascular and neuronal conditions and the role they play in other, non-excitable tissues is beginning to emerge.
The Importance of the Ca2+ Ion as a Signaling Molecule
In most cells the intracellular concentration of ionized calcium ([Ca2+]in) is actively kept very low, at about 100-200 nM, while the plasma Ca2+ concentration ([Ca2+]out) is at about 2 mM. This is achieved by several mechanisms. Firstly, the lipid membrane is impermeable to Ca2+, unless a Ca2+ conducting channel in the plasma membrane is open, allowing ion diffusion along its electrochemical gradient. Other mechanisms include fast Ca2+ buffering either by active transport into organelles such as mitochondria and endoplasmic reticulum (ER) or by binding to cytosolic proteins. Therefore, small and local increases in [Ca2+]in trigger the cellular signaling cascades which underlie the basis of many physiological systems1.
This paper focuses on a set of plasma membrane proteins that respond to membrane potential depolarization by opening a Ca2+ selective pore: the voltage dependent/gated/operated Ca2+ (CaV) channels.
Structure and Naming of CaV Channels
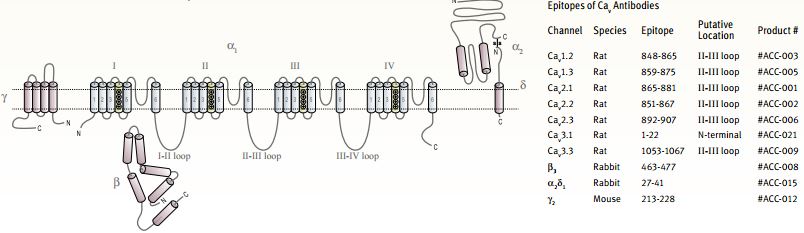
The principal protein (α1 subunit) that forms a CaV channel is a large (~2000 amino acids) protein that spans the membrane 24 times and includes the Ca2+ selective pore, four voltage sensors and large intracellular loops with different regulatory functions. Such proteins are a single polypeptide analogous to the archetypal KV channel tetramer (CaV proteins contain four homologous domains, each resembles a KV subunit)2. Recently, the structures of purified skeletal and cardiac muscle L-type channels was determined by electron microscopy at low resolution3-5.
In addition, three other protein families are referred to as CaV auxiliary subunits: a cytoplasmic β subunit, a membrane anchored extracellular α2δ subunit and the integral membrane γ subunit. All the auxiliary subunits affect the activity of all pore forming α1 subunits differently and to different degrees6-9. In 2004, the high resolution crystal structures of the core domain of three β subunits as well as of β subunits complexed with part of the CaV α1 subunit, were published, shedding light on the intersubunit relationship within a CaV complex10-12,13.
The ten CaV α1 subunit genes are divided into three subfamilies on both a functional and sequence homology basis (Table). The CaV1 subfamily includes four genes that encode L-type channels and the CaV3 subfamily includes three genes that encode T-type channels. The CaV2 subfamily contains three isoforms encoding P/Q- , N- and R-type channels, with all three contributing to neurotransmitter release in neurons14.
The Role of Voltage Activation
The control of Ca2+ entry into the cell by selective pathways, which sense the voltage of the plasma membrane, enables fast, localized and brief transduction of electrical signal- into cytosolic Ca2+– signal. Hence, the four voltage sensors in each CaV channel detect action potentials (and many other voltage signals) and induce a conformational change which allows Ca2+ ions to permeate into the cell resulting in a brief Ca2+ influx. This elevation of the Ca2+ concentration near the channel might activate any Ca2+ dependent enzyme or processes, which senses it. Two examples which illustrate such very localized signaling are synaptic transmission and calmodulin modulation of channel function.
The process of neurotransmitter release from neuronal nerve terminals is triggered by action potential invasion of the terminal leading to opening of CaV channels. CaV2 subfamily channels are localized in nerve terminals in the vicinity of docked synaptic vesicles which contain the neurotransmitter. Upon fusion of the vesicles with the plasma membrane the neurotransmitter is released. The “fusion machinery” consists of a number of SNARE proteins (on both the plasma and vesicle membranes), among them Ca2+ dependent proteins. These are activated directly by Ca2+ entering via CaV channels, which are already complexed with parts of the “machinery” such as syntaxin15,16. Such a process highlights the critical role played by CaV channels in the transformation of an electrical signal in one cell (or cell population) into a chemical signal affecting neighboring cells (synaptic transmission) and remote cells or organs (hormone secretion).
Several CaV channels were shown to have an inactive (Ca2+ free) calmodulin tethered to a cytoplasmic C-terminus of the protein. Once the channel opens in response to membrane potential depolarization, the tethered calmodulin binds the entering Ca2+, is activated, and in many cases enforces channel closure (Ca2+dependent inactivation), forming a negative feedback loop on the Ca2+ entry activity17.
Tissue Distribution
Although all CaV channels are mostly studied in excitable tissues, i.e. nerve and muscle, their presence and roles in other tissues (such as hormonal glands,18,19 immune system cells,20 kidney,21 tumors22 etc.) are important in a physiological and pathological context. CaV channels control key points in the Ca2+ activation of processes such as neurotransmitter release, skeletal, cardiac and smooth muscle contraction, gene expression, apoptosis and proliferation.
Some CaV specific isoforms or splice variants are to a large extent cell type specific. For example, CaV1.1 is expressed almost specifically in skeletal muscle and the protein functions as the main L-type channel in these cells, while the CaV1.4 isoform is expressed mainly in the retina23 and T-lymphocytes20 and underlies their L-type currents. In contrast, CaV1.2 is widely expressed, and this gene product underlies L-type currents in both cardiac and smooth muscle myocytes, in many neurons and pancreatic β cells. However, different gene products (splice variants) of the CaV1.2 gene might be selectively expressed in one tissue compared to another24,25. CaV1.3 is also expressed in several different tissues and knockout mice are affected with hearing loss and cardiac arrhythmia26 as well as with impaired glucose metabolism27.
The CaV2 family’s most notable function is in linking membrane potential depolarization to neurotransmitter release in presynaptic nerve terminals. As such, these channels are key regulators of neuronal communication. Within various nerve cell populations, CaV2 family members are differentially expressed, sometimes with overlapping patterns28. Here as well, splice variants may be specifically expressed. A most interesting example is related to pain control with regard to an N-type channel (CaV2.2) variant, which is selectively expressed in sensory neurons in DRG29. CaV2 channels are modulated by G-protein coupled receptors, a cellular pathway that is most important to physiological processes such as hormonal regulation of neuronal activity as well as in clinical interventions such as morphine-induced analgesia30.
The three T-type (CaV3.1-3) channel isoforms are also widely and differentially distributed and for members of this subfamily as well specific expression of splice variants was demonstrated31.
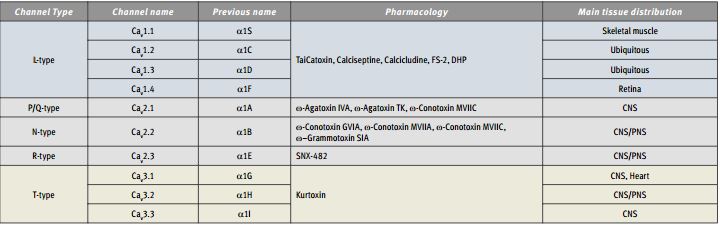
Immunohistochemical staining of Syntaxin 2 with Anti-Syntaxin 2 Antibody (#ANR-008) in the rat cerebellum. The soma of Purkinje cells are stained, as well as the upper layers of the cerebellum. Immunoreactive product is black.
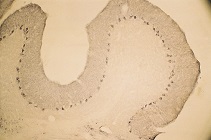
1. Anti-Syntaxin 4 Antibody (#ANR-004) (1:600).
2. Anti-Syntaxin 4 Antibody, preincubated with the negative control antigen.
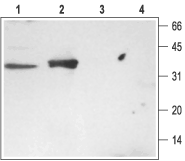
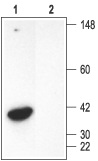
1. Rat brain membranes, Anti-Syntaxin 3 Antibody (#ANR-005) (1:200).
2. Rat kidney membranes, Anti-Syntaxin 3 Antibody (1:200)3. Rat brain membranes, Anti-Syntaxin 3 Antibody, preincubated with the negative control antigen.4. Rat kidney membranes, Anti-Syntaxin 3 Antibody, preincubated with the negative control antigen.
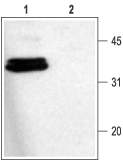
1. Anti-Syntaxin 1 Antibody (#ANR-002) (1:1000).
2. Anti-Syntaxin 1 Antibody, preincubated with the control fusion protein antigen.
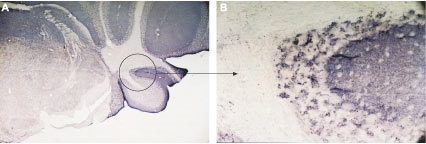
Immunohistochemical staining using Anti-VAMP-2 Antibody (#ANR-007) in mouse cerebellum (A). The entire molecular layer and patches of the granule layer were stained. The patchy pattern of staining is demonstrated in the circle shown in (A), magnified in (B).
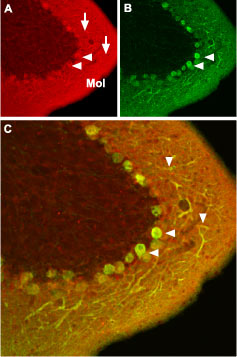
Immunohistochemical staining of CaV1.2 channel with Anti-CaV1.2 (CACNA1C) Antibody (#ACC-003) in mouse cerebellum. A. CaV1.2 channel (red) appears in Purkinje cells (horizontal arrows) and is distributed diffusely in the molecular layer (Mol) including in Purkinje dendrites (vertical arrows). B. staining of Purkinje nerve cells with mouse anti calcium binding protein (green) in the section demonstrates the location of dendrites in the molecular layer. C. Confocal merge of CaV1.2 and CBD28K.
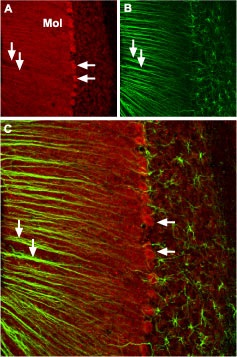
Immunohistochemical staining of CaV2.1 channel with Anti-CACNA1A (CaV2.1) Antibody (#ACC-001) in mouse cerebellum.
A. CaV2.1 channel (red) appears in Purkinje cells (horizontal arrows) and is distributed diffusely in the molecular layer (Mol) including in astrocytic fibers (vertical arrows). B. Staining of astrocytic fibers with glial fibrillary acidic protein (blue – originally green digitally edited to blue) in the section demonstrates the location of astrocytic fibers in the molecular layer. C. Confocal merge CaV2.1 and GFAP.
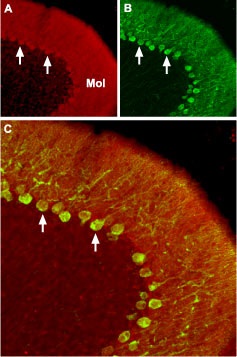
Immunohistochemical staining of CaV2.2 channel with Anti-CACNA1B (CaV2.2) Antibody (#ACC-002) in mouse cerebellum.A. CaV2.2 channel (red) appears in Purkinje cells (arrows) and is distributed diffusely in the molecular layer (Mol). B. staining of Purkinje nerve cells with mouse anti calcium binding protein (green) demonstrates the restriction of CaV2.2 to cell bodies but not to dendrites in the molecular layer. C. Confocal merge of CaV2.2 and CBD28K.

1. Anti-CaV1.2 (CACNA1C) Antibody (#ACC-003) (1:200).
2. Anti-CaV1.2 (CACNA1C) Antibody, preincubated with the negative control antigen.
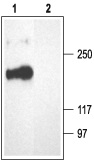
1. Anti-CACNA1A (CaV2.1) Antibody (#ACC-001) (1:200).
2. Anti-CACNA1A (CaV2.1) Antibody, preincubated with the negative control antigen.

1. Anti-CACNA1B (CaV2.2) Antibody (#ACC-002) (1:200).
2. Anti-CACNA1B (CaV2.2) Antibody, preincubated with the negative control antigen.
Channelopathies, Toxins and Clinical CaV Channel Blockers
Mutations in CaV channel genes are linked to several inherited neuronal (CaV2.1) and muscular diseases (CaV1.1)32 as well to a multisystemic syndrome involving autism (CaV1.2)33. The latter exemplifies the general role played by this L-type channel in regulating Ca2+ metabolism in excitable as well in non-excitable tissues (such as bone and lung). Although these mutations are rare, pharmacological interventions that target CaV channels are widely used in the clinic,34-37 among them antihypertensive drugs acting on smooth muscle CaV1.2 channels or anticonvulsant drugs that target brain T-type channels. Recently, the FDA has approved PrialtTM (Ziconotide) a synthetic version of ω-Conotoxin MVIIA, as a painkiller for some indications38,39.
One of the first methods used to differentiate among voltage dependent Ca2+ channels in assessing their particular physiological roles, is based on differential pharmacological sensitivities. Particularly, the differentiation between the presynaptic CaV2 subfamily isoforms was facilitated due to specific toxin sensitivities. The cone snail peptides ω-Conotoxin GVIA and ω-Conotoxin MVIIA40 inhibit the CaV2.2 channel potently and specifically, while the spider toxin ω-Agatoxin IVA41 is specific for CaV2.1. Later, the tarantula peptide, SNX-48242 was shown to be a specific blocker of CaV2.3, which was associated with the toxin resistant R-type current. These toxins were used to block transmission in different synapses43-45.
Other compounds that block CaV channels are very useful in differentiating between subfamilies but have little selectivity toward specific isoforms within the subfamily. The CaV1 subfamily proteins are often referred to as DHP receptors, as this group of chemical compounds interacts specifically with CaV1 channels either as antagonists (such as Nimodipine) or agonists (i.e. (S)(-)-Bay K8644). Other examples include, the scorpion peptide kurtoxin, that blocks all three T-type channels with similar potencies, but has little effect on other CaV channels46,47. In a similar manner Calcicludine48,49 and Calciseptine50,51, toxins purified from the venom of the green and black mamba respectively, block L-type channels specifically, but little is known regarding their selectivity between CaV1 subfamily channels.
Acknowledgements: We thank Annette C. Dolphin, Ph.D. from Department of Pharmacology, UCL, UK for her helpful comments.

Staining of CaV1.3 in adult rat dorsal root ganglion (DRG) with Anti-CaV1.3 (CACNA1D) Antibody (#ACC-005). Staining (black) appeared mostly in clusters of cells (arrows) with variable intensity, within the ganglion; not in the roots.

1. Anti-CaV1.3 (CACNA1D) Antibody (#ACC-005) (1:200).2. Anti-CaV1.3 (CACNA1D) Antibody, preincubated with the negative control antigen.
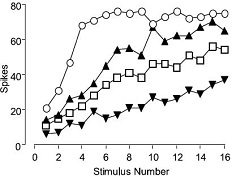
Dose-related effects of SNX-482 (#RTS-500)on the wind-up of spinal neurons in Spinal Nerve Ligated (SNL) rats. The number of action potentials evoked per stimulus were plotted against the stimulus number, before and after drug administration in a single dorsal horn neuron. SNX-482 produced dose-related inhibitions of the wind-up of spinal neurons in SNL rats.
Adapted from Stephens, G.J. Matthews, E. & Dickenson, A.H. (2003) Soc. for Neurosci. Abst. 33:589.10 Dept. of Pharmacology, University College London, UK.

The effect of 5 μM (±)-Bay K8644 (#B-350) on heterologously expressed L-type Ca2+ currents (CaV1.2 / α2δ1 / β2a, RNA injected into Xenopus oocytes). Left: I-V relation before (diamonds) and during (circles), bath perfusion of the compound. Right: Current response to 200 ms depolarization to +20 mV (from holding potential of -100 mV) before (blue) and during (green) perfusion of the drug.
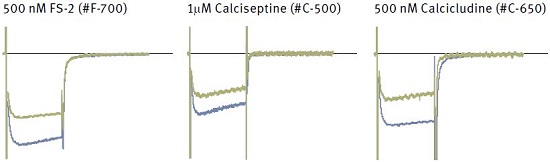
Each panel shows superimposed currents before (blue) and during bath perfusion of the indicated toxin (green).
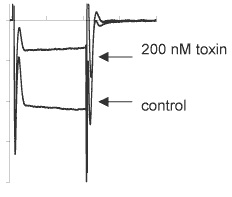
Inhibition of CaV2.2 channels expressed in Xenopus oocytes by ω-Conotoxin MVIIA (#C-670). Traces before or during perfusion of 200 nM toxin.
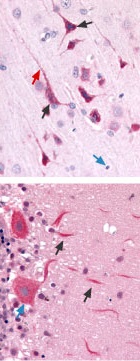
Immunohistochemical staining of CaV3.3 channel with Anti-CaV3.3 (CACNA1I) Antibody(#ACC-009) in in rat brain cortex and cerebellum. (A) Picture showing the third layer of the brain cortex. Pyramidal neurons cells (black arrow) and their axons (green arrow) shows strong staining. However, glial cells (blue arrows) show no staining at all. (B) Picture showing the Purkinje layer of the rat cerebellum. Note that Purkinje cells (blue arrows) and their axons (black arrows) were strongly stained. Staining product is red and counterstain is hematoxylin.
Immunohistochemistry data provided by LifeSpan Biosciences, Seattle, USA.

1. Anti-CaV3.3 (CACNA1I) Antibody (#ACC-009) (1:200).
2. Anti-CaV3.3 (CACNA1I) Antibody, preincubated with the negative control antigen.
References
- Berridge, M.J. et al. (2003) Nat. Rev. Mol. Cell Biol. 4, 517.
- Yu, F.H. and Catterall, W.A. (2004) Sci. STKE 2004, re15.
- Wang, M.C. et al. (2002) J. Mol. Biol. 323, 85.
- Wolf, M. et al. (2003) J. Mol. Biol. 332, 171.
- Wang, M.C. et al. (2004) J. Biol. Chem. 279, 7159.
- Yasuda, T. et al. (2004) Euro. J. Neurosci. 20, 1.
- Dolphin, A.C. (2003) J. Bioenerg. Biomembr. 35, 599.
- Canti, C. et al. (2003) Current Neuropharmacology 1, 209.
- Arikkath, J. and Campbell, K.P. (2003) Curr. Opin. Neurobiol. 13, 298.
- Opatowsky, Y. et al. (2004) Neuron 42, 387.
- Chen, Y.H. et al. (2004) Nature 429, 675.
- Van Petegem, F. et al. (2004) Nature 429, 671.
- Richards, M.W. et al. (2004) Trends Pharmacol. Sci. 25, 626.
- Catterall , W.A. et al. (2003) Pharmacol. Rev. 55, 579.
- Atlas, D. (2001) J. Neurochem. 77, 972.
- Spafford, J.D. and Zamponi, G.W. (2003) Curr. Opin. Neurobiol. 13, 308.
- Liang, H. et al. (2003) Neuron 39, 951.
- Yang, S.N. and Berggren P.O. (2005) Am. J. Physiol. Endocrinol. Metab. 288, E16.
- Sedej, S. et al. (2004) J. Physiol. 555, 769.
- Kotturi, M.F. et al. (2003) J. Biol. Chem. 278, 46949.
- Jensen, B.L. et al. (2004) Nephrol. Dial. Transplant. 19, 1368.
- Wang, X.T. et al. (2000) Am. J. Pathol. 157, 1549.
- McRory, J.E. et al. (2004) J. Neurosci. 24, 1707.
- Liao, P. et al. (2004) J. Biol. Chem. 279, 50329.
- Tang, Z.Z. et al. (2004) J. Biol. Chem. 279, 44335.
- Platzer, J. et al. (2000) Cell 102, 89.
- Namkung, Y. et al. (2001) J. Clin. Invest. 108, 1015.
- Meir, A. et al. (1999) Physiol. Rev. 79, 1019.
- Bell, T.J. et al. (2004) Neuron 41, 127.
- Dolphin A.C. (2003) Pharmacol. Rev. 55, 607.
- Perez-Reyes, E. (2003) Physiol. Rev. 83, 117.
- Meir, A. and Dolphin A.C. (2002) Modulator 16, 4.
- Splawski, I. et al. (2004) Cell 119, 19.
- Triggle, D.J. (2003) Assay Drug Dev. Technol. 1, 719.
- Kochegarov, A.A. (2003) Cell Calcium 33, 145.
- Elmslie, K.S. (2004) J. Neurosci. Res. 75, 733.
- Rogawski, M.A. and Löscher, W. (2004) Nat. Rev. Neurosci. 5, 553.
- Bogin, O. (2004) Modulator 19, 14.
- Gohar, O. (2004) Modulator 19, 9.
- Terlau, H. and Olivera, B.M. (2004) Physiol. Rev. 84, 41.
- Adams, M.E. (2004) Toxicon 43, 509.
- Newcomb, R. et al. (1998) Biochemistry 37, 15353.
- Kerr, L.M. and Yoshikami, D. (1984) Nature 308, 282.
- Wang, G. et al. (1999) J. Neurosci. 19, 9235.
- Stephens, G.J. et. al. (2001) Eur. J. Neurosci. 13, 1902.
- Chuang, R.S. et al. (1998) Nat. Neurosci. 1, 668.
- Sidach S.S. and Mintz, I.M. (2002) J. Neurosci. 22, 2023.
- Schweitz, H. et al. (1994) P.N.A.S. 91, 878.
- Stotz, S.C. et al. (2000) J. Membr. Biol. 174, 157.
- Schweitz, H. et al. (1990) Toxicon 28, 847.
- De Weille, J.R. et al. (1991) P.N.A.S. 88, 2437.