Overview
- Peptide (C)NGVPESTSTDTPPDIDLHN, corresponding to amino acid residues 392-410 of human Kir2.1 (Accession P48049). Intracellular, C-terminus.
- Western blot analysis (1:500-1:1000).
 Western blot analysis of mouse heart lysate (lanes 1 and 3) and rat brain membranes (lanes 2 and 4):1-2. Guinea pig Anti-Kir2.1/KCNJ2 Antibody (#APC-026-GP), (1:500).
Western blot analysis of mouse heart lysate (lanes 1 and 3) and rat brain membranes (lanes 2 and 4):1-2. Guinea pig Anti-Kir2.1/KCNJ2 Antibody (#APC-026-GP), (1:500).
3-4. Guinea pig Anti-Kir2.1/KCNJ2 Antibody, preincubated with Kir2.1/KCNJ2 Blocking Peptide (#BLP-PC026).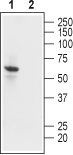 Western blot analysis of human U-87 MG glioblastoma cell lysate:1. Guinea pig Anti-Kir2.1/KCNJ2 Antibody (#APC-026-GP), (1:500).
Western blot analysis of human U-87 MG glioblastoma cell lysate:1. Guinea pig Anti-Kir2.1/KCNJ2 Antibody (#APC-026-GP), (1:500).
2. Guinea pig Anti-Kir2.1/KCNJ2 Antibody, preincubated with Kir2.1/KCNJ2 Blocking Peptide (#BLP-PC026).- Following a broad screen of secondary antibodies, the following was used for this application: #106-035-006 (Jackson ImmunoResearch).
Kir2.1 is a member of the family of inward rectifying K+ channels. The family includes 15 members that are structurally and functionally different from the voltage-dependent K+ channels.1
The family’s topology consists of two transmembrane domains that flank a single and highly conserved pore region with intracellular N- and C-termini. As is the case for the voltage-dependent K+ channels the functional unit for the Kir channels is composed of four subunits that can assemble as either homo- or heterotetramers.
Kir channels are characterized by a K+ efflux that is limited by depolarizing membrane potentials thus making them essential for controlling resting membrane potential and K+ homeostasis.
Kir2.1 is a member of the Kir2.x subfamily that includes four members (Kir2.1- Kir2.4) that are characterized by strong inward rectification and high constitutive activity.
Kir2.1 is expressed in a variety of tissues including the heart, brain, vascular smooth muscle cells and skeletal muscles.
In the heart, Kir2.1 is a molecular component of the IK1 current that is responsible for setting the resting membrane potential, preventing membrane hyperpolarization due to Na+ pump activity, influencing propagation velocity, altering the electrical space constant, and promoting late phase repolarization.2 In fact, mutations in Kir2.1 channels have been linked to a form of long QT syndrome (LQT7) known as Andersen's syndrome that is characterized by cardiac arrhythmias, periodic paralysis, and dysmorphic features.3
