Overview
- Peptide (C)HGDLLELGPPANHT, corresponding to amino acid residues 93-106 of rat Kir4.1 (Accession P49655). Extracellular loop.
- Rat and mouse brain lysates (1:500-1:2000).
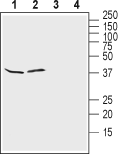 Western blot analysis of rat brain lysates (lanes 1 and 3) and mouse brain lysates (lanes 2 and 4):1, 2. Anti-Kir4.1 (KCNJ10) (extracellular) Antibody (#APC-165), (1:500).
Western blot analysis of rat brain lysates (lanes 1 and 3) and mouse brain lysates (lanes 2 and 4):1, 2. Anti-Kir4.1 (KCNJ10) (extracellular) Antibody (#APC-165), (1:500).
3, 4. Anti-Kir4.1 (KCNJ10) (extracellular) Antibody, preincubated with Kir4.1/KCNJ10 (extracellular) Blocking Peptide (#BLP-PC165).
Rat brain sections (1:200), (Cui, Y. et al. (2018) Nature 554, 323.).
- Human U-87 MG glioblastoma cell line (1:100).
Kir4.1 is a member of the inward rectifying K+ channel family. The family includes 15 members that are structurally and functionally different from the voltage-dependent K+ channels.
The family’s topology consists of two transmembrane domains that flank a single and highly conserved pore region with intracellular N- and C-termini. As is the case for the voltage-dependent K+ channels the functional unit for the Kir channels is composed of four subunit that can assembly as either homo or heteromers.
Kir channels are characterized by a K+ efflux that is limited by depolarizing membrane potentials thus making them essential for controlling resting membrane potential and K+ homeostasis.
Kir4.1 is a member of the Kir4 subfamily that includes one other member: Kir4.2. Kir4.1 can co-assemble with Kir4.2 but also with other Kir channels such as Kir2.1 and Kir5.1.
The Kir4 subfamily has been classified as weak rectifiers with intermediate conductance.
Kir4.1, encoded by KCNJ10, is mainly expressed in brain, specifically in glia cells, but also in retina, ear and kidney.1,2
It has been proposed that Kir4.1 has an essential role in glial K+ buffering, a process that re-uptakes the K+ released during neuronal activity into the intracellular interstitial space. Loss of Kir4.1 causes retinal defects and loss of endochoclear potential.3
